Oceans are a rich source of structurally unique bioactive compounds from the perspective of potential therapeutic agents. Marine peptides are a particularly interesting group of secondary metabolites because of their chemistry and wide range of biological activities. Among them, cyclic peptides exhibit a broad spectrum of antimicrobial activities, including against bacteria, protozoa, fungi, and viruses. Moreover, there are several examples of marine cyclic peptides revealing interesting antimicrobial activities against numerous drug-resistant bacteria and fungi, making these compounds a very promising resource in the search for novel antimicrobial agents to revert multidrug-resistance.
- marine peptides
- cyclic peptides
- cyclic depsipeptides
- antimicrobial resistance
- peptide synthesis
1. Introduction
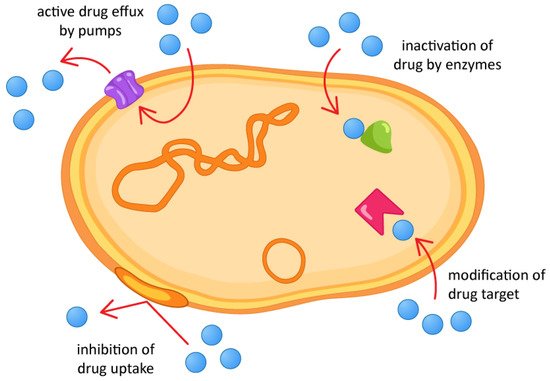
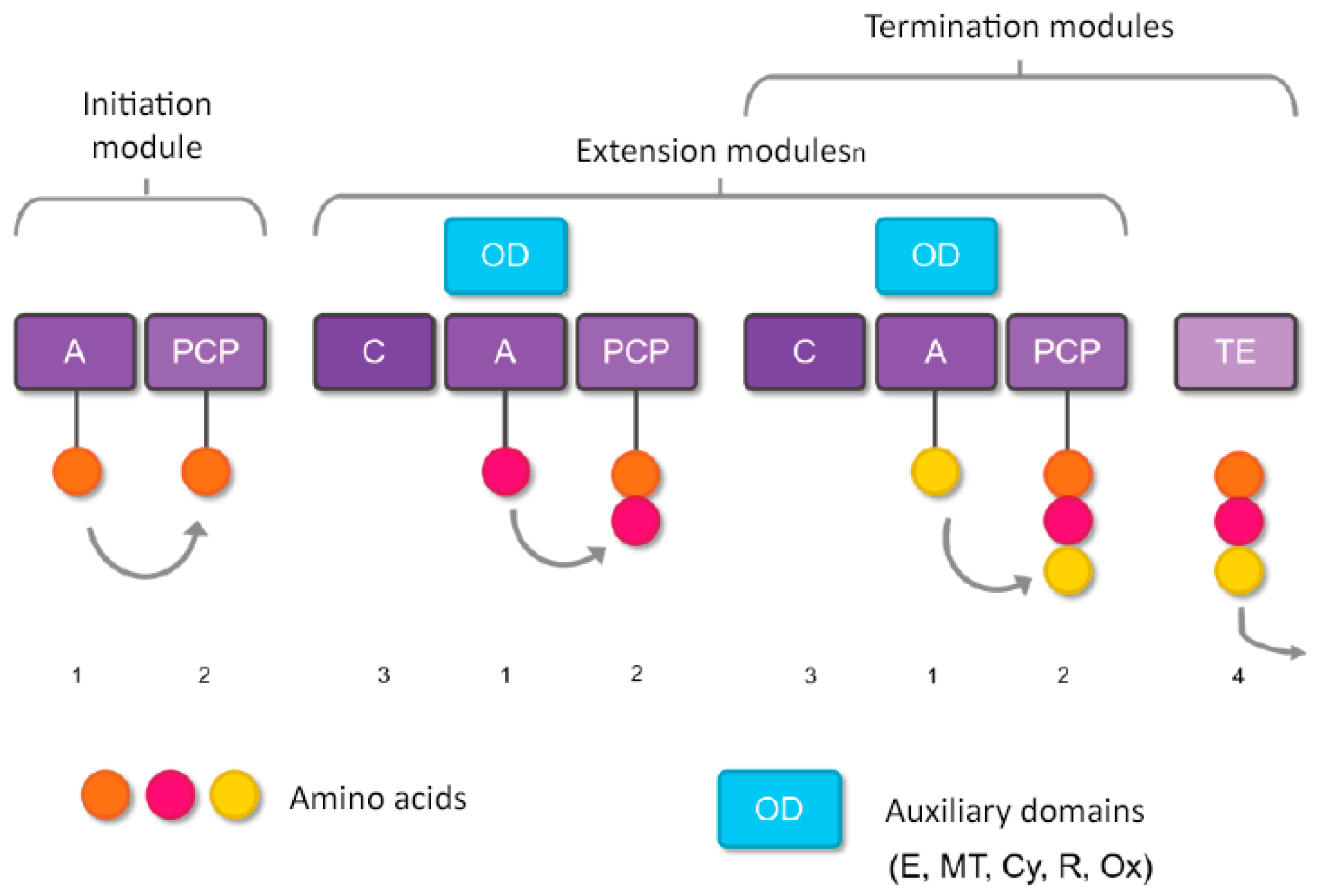
2. Marine Cyclic Peptides with Antimicrobial Activities
2.1. Sponge-Produced Cyclic Peptides
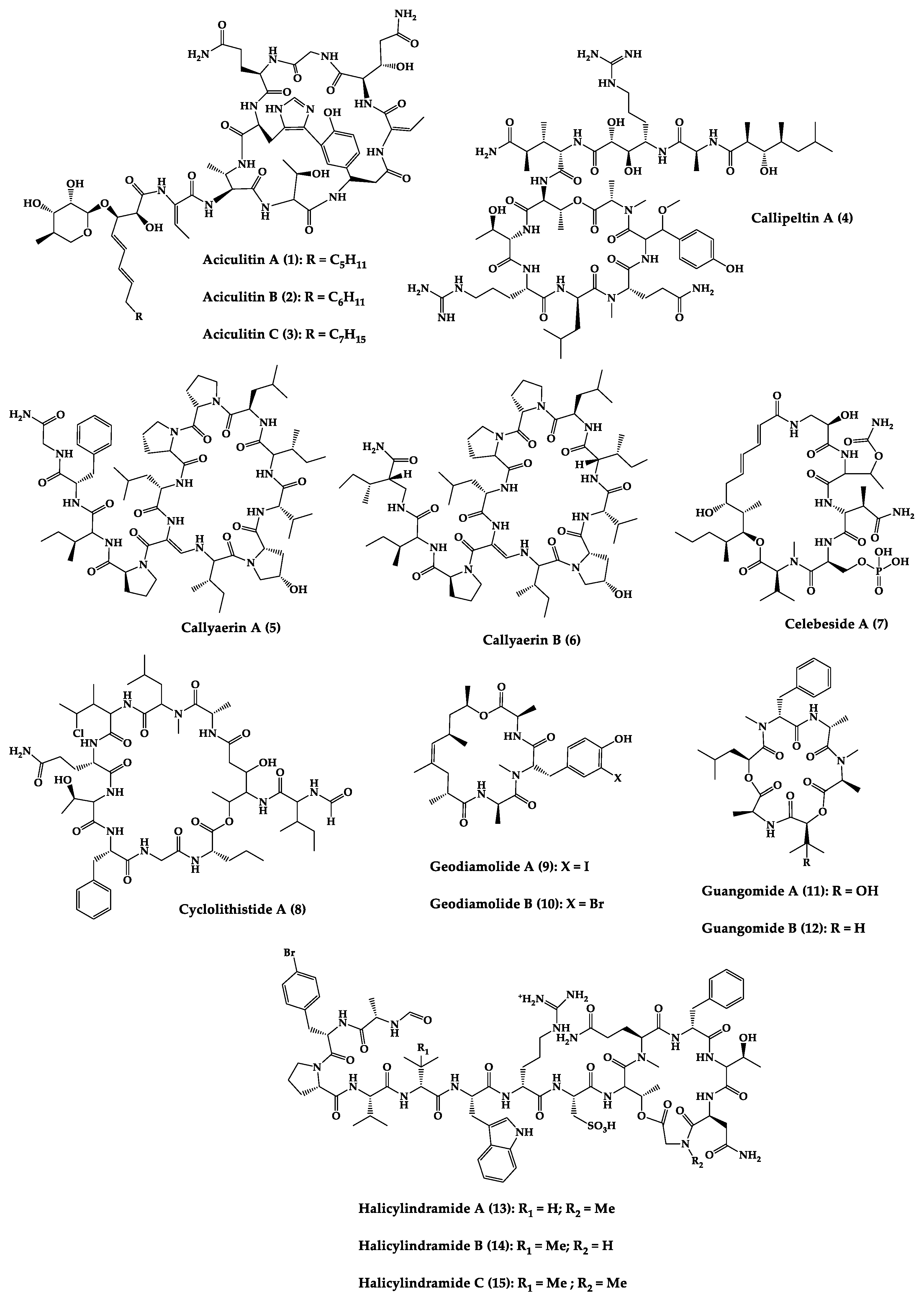
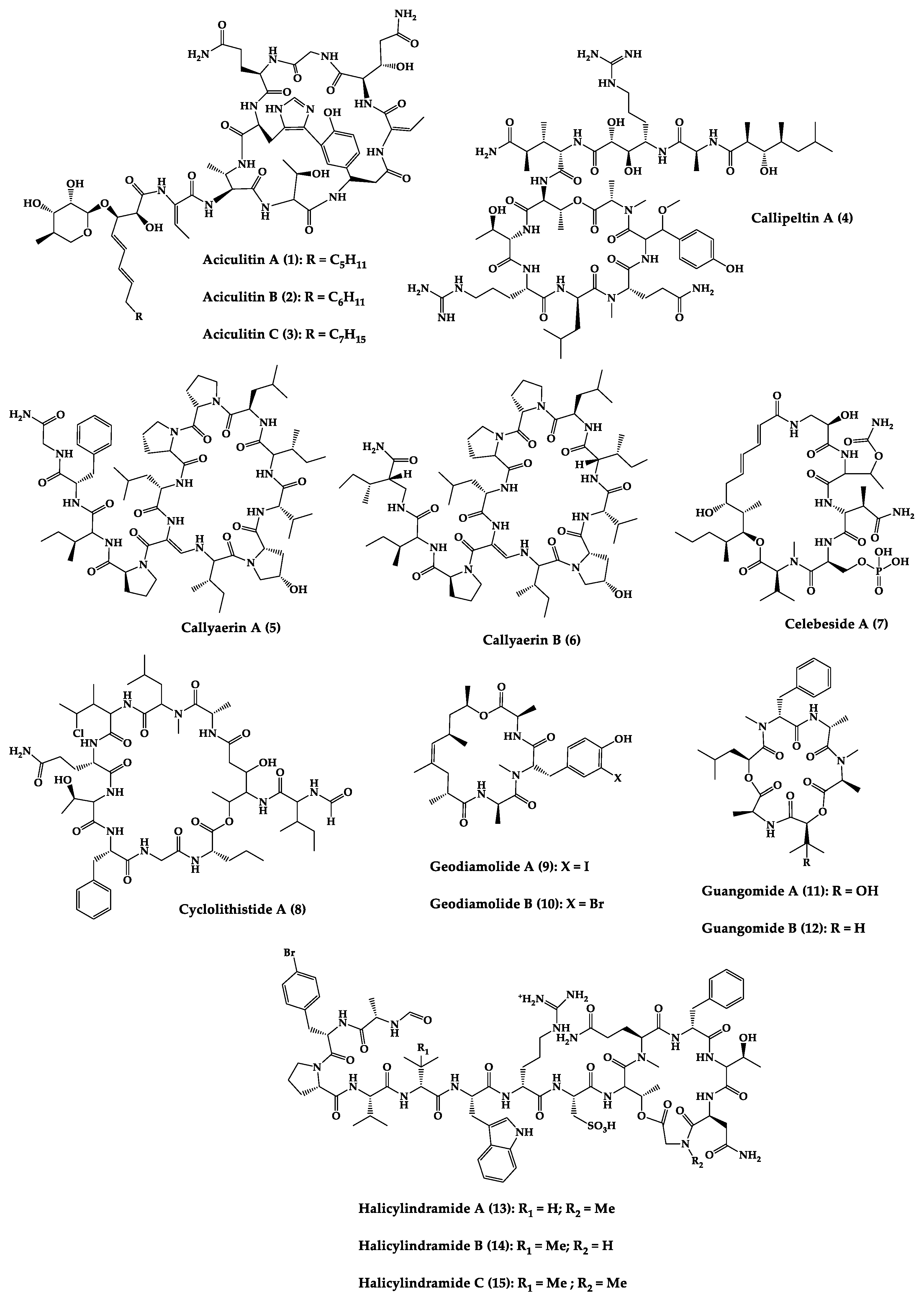
| Compound | Structure | Source | Antimicrobial Activity | Synthesis | References |
|---|---|---|---|---|---|
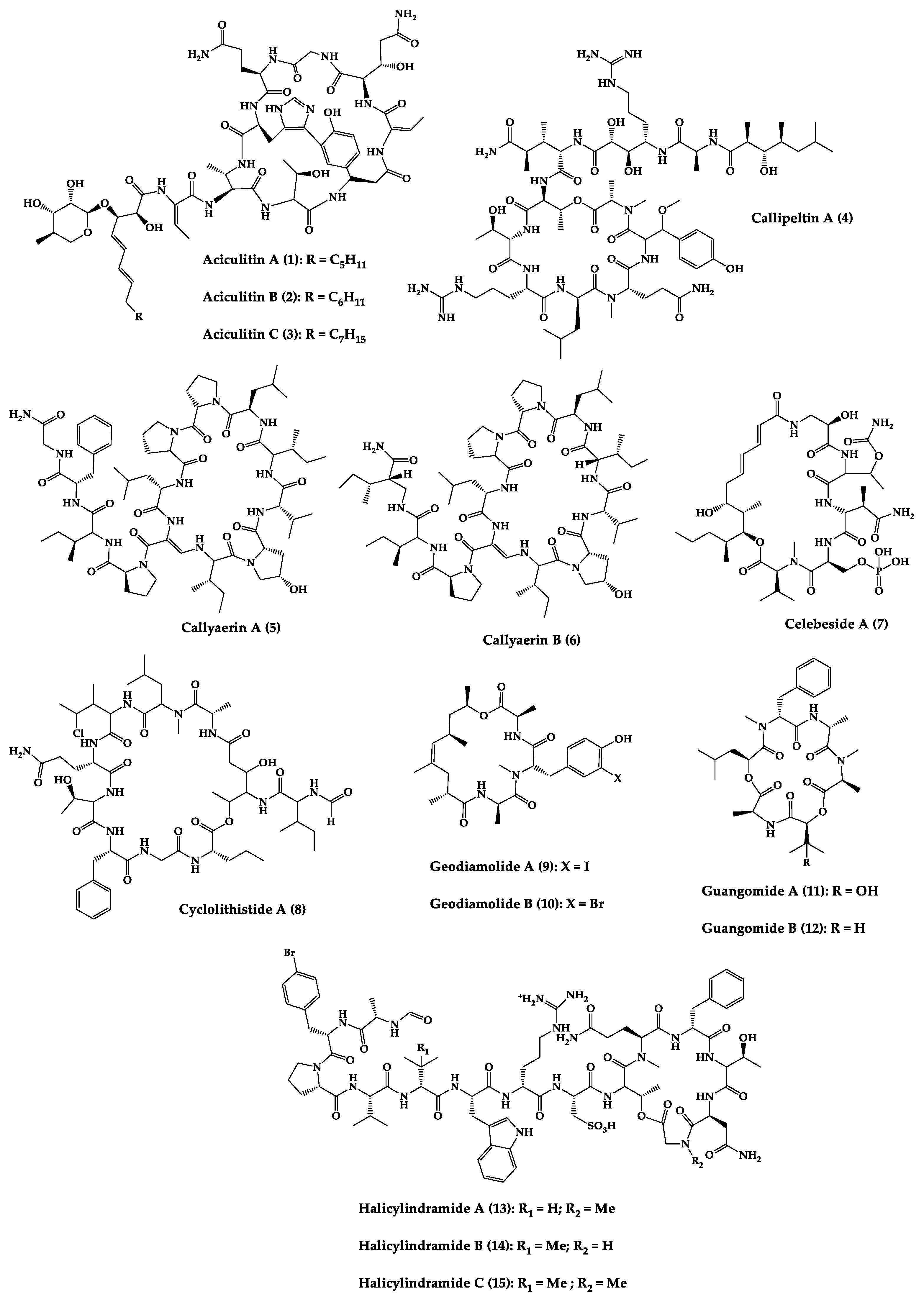
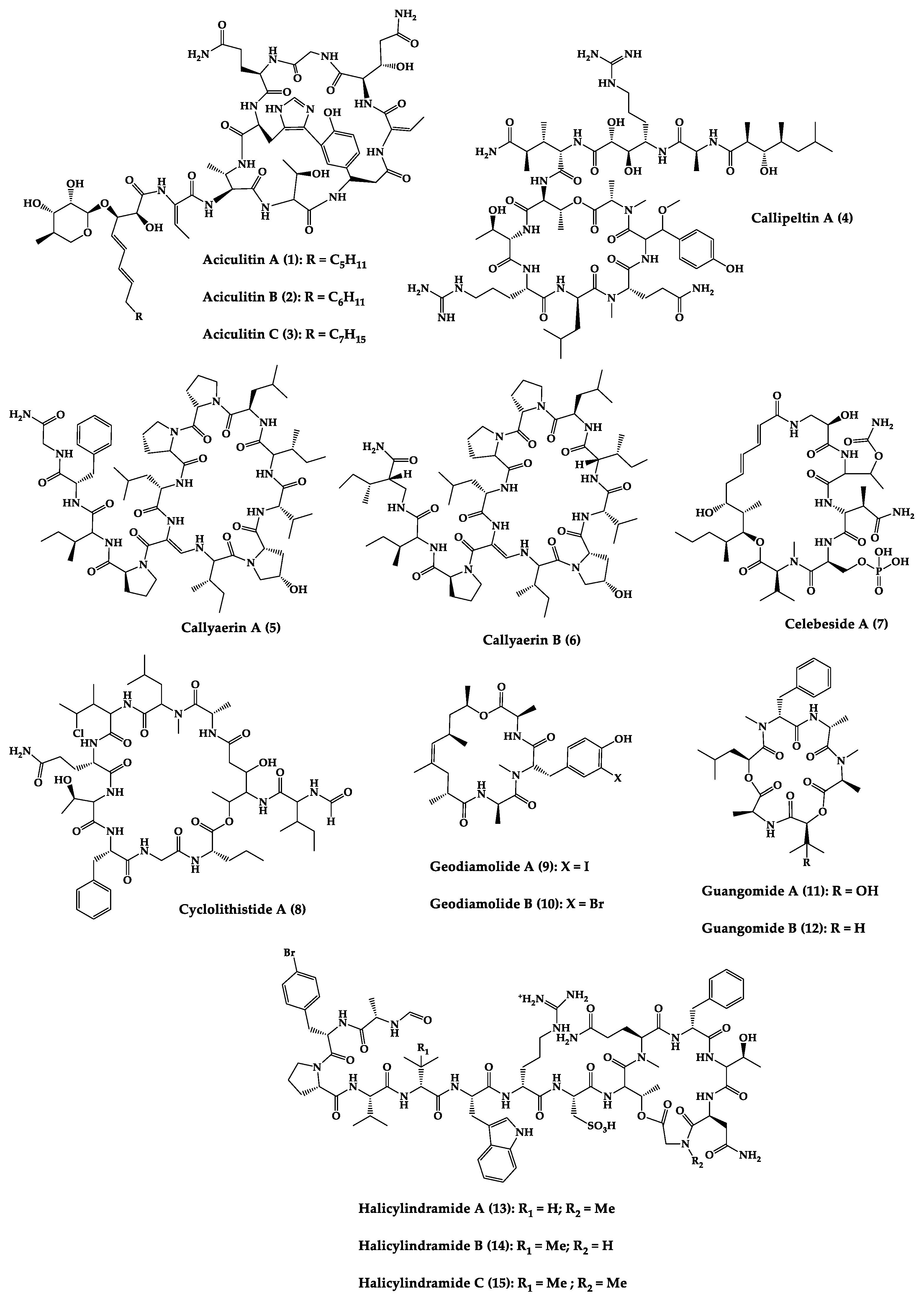
| Aciculitins A-C (1–3) | Bicyclic octa-peptides | Aciculites orientalis | C. albicans (2.5 µg/disk, standard disk assay) | Semi-synthesis | [124][125] |
| Callipeltin A (4) | Cyclic deca-depsipeptide | Callipelta sp. | HIV-1 infection inhibition (CD50 = 0.29 µg/mL, ED50 = 0.01 µg/mL), C. albicans (100 µg/disk) | Total synthesis of analogues | [126][127][128][129][130] |
| Callyaerins A (5) and B (6) | Cyclic undeca-peptides | Callyspongia aerizusa | IC90: M. tuberculosis (2 μM and 5 μM, respectively), isoniazide (0.625 μM) | Total synthesis | [131][132] |
| Celebeside A (7) | Cyclic penta-depsipeptide | Siliquaria-spongia mirabilis | IC50: Neutralized HIV-1 (1.9 µg/mL) | - | [123] |
| Cyclolithistide A (8) | Cyclic deca-despipeptide | Bacteria symbiosis Theonella swinhoei | C. albicans (20 µg/disk) | - | [133] |
| Geodiamolides A (9) and B (10) | Cyclic depsipeptides | Geodia sp. | MIC: C. albicans (31.3 µg/mL) | Total synthesis | [134][135][136] |
| Guangomides A (11) and B (12) | Cyclic tetra-depsipeptides | Unidentifiable sponge derived fungus | MIC: S. epidermidis (100 µg/mL), E. durans (100 µg/mL) |
- | [137] |
| Halicylindramides A-C (13–15) |
Cyclic tetra-decapeptides | Halichondria cylindruta | M. ramanniana (7.5 µg/disk) | Total synthesis and analogues | [108][138][139] |
| Homophymine A (16) | Cyclic undeca-depsipeptide | Homophymia sp. | IC50: HIV-1 infection cytoprotective (75 nM) | Semi-synthesis | [109][140][141] |
| Hymenamides A (17), B (18), C (19), and E (20) | Cyclic hepta-peptides | Hymeniacidon sp. | MIC: C. albicans (33–66 µg/mL), C. neoformans (33–133 µg/mL) | Total synthesis and analogues | [142][143][144] |
| Jasplakinolide (or jaspamide) (21) | Cyclic depsipeptide | Jaspis sp. | H. virescens (LC50 = 4 ppm), N. brasiliensis (LD50 < 1 µg/mL), C. albicans (MIC > 25 µg/mL), in vivo murine vaginal C. albicans infection (2% jasplakinolide was equivalent in efficacy to administration of miconazole nitrate at 2%) |
Total synthesis and analogues | [110][111][112][145][146][147] |
| Koshikamides F (22) and H (23) | Cyclic heptadeca-peptides | Theonella swinhoei and T. cupola | IC50: HIV-1 neutralization (2.3–5.5 µM) | - | [45][113] |
| Microcionamides A (24) and B (25) | Cyclic hexapeptides | Clathria abietina | MIC: M. tuberculosis (5.7 µM) |
- | [148] |
| Microsclero-dermins A–K (26–36) and anhydromicros-clerodermin C (37) |
Cyclic hexapeptides | Cyanobacteria simbiosis Microsclero-derma herdmani sp. and Theonella sp. | C. albicans (2.5–100 µg/disk, standard disk assay) |
Total synthesis and analogues | [149][150][151][152][153] |
| Microspinosamide (38) | Cyclic trideca-depsipeptide | Sidonops microspinosa | EC50: HIV-1 infection inhibition (0.2 µg/mL) |
Semi-synthesis | [114][154] |
| Mirabamides A–H (39–46) |
Cyclic glyco-depsipeptides | Siliquarias-pongia mirabilis and Stelletta clavosa | IC50: neutralized and fusion HIV-1 (40 nM–3.9 µM), B. subtilis, C. albicans (1–5 µg/disk) |
Semi-synthesis | [115][116][155] |
| Nagahamide A (47) | Cyclic hexa-depsipeptide | Theonella swinhoei | E. coli or S. aureus (50 µg/disk, inhibition zone 7 mm) |
Semi-synthesis | [156][157] |
| Neamphamide A (48) | Cyclic undeca-depsipeptide | Neamphius huxleyi | EC50: HIV-1 infection cytoprotective (28 nM) | - | [118] |
| Neamphamide B (49) | Cyclic undeca-depsipeptide | Neamphius sp. | MIC: M. smegmatis (1.56 µg/mL), M. bovis (6.2–12.5 µg/mL) |
- | [119] |
| Neosiphoniamolide A (50) | Cyclic tetra-depsipeptide | Neosiphonia suprtes | P. oryzae (IC90 = 5 ppm) H. gramineum (MIC ≤ 2 µg/mL) | - | [158] |
| Papuamides A (51) and B (52) | Cyclic depsipeptides | Bacteria symbiosis Theonella mirabilis and Theonella swinhoei | EC50: HIV-1 infection inhibition (1–74 ng/mL) |
Total synthesis and analogues | [120][155][159][160][161][162] |
| Polydiscamide A (53) | Cyclic tridecapeptide | Discodermia sp. | MIC: B. subtilis (3.1 µg/mL) | Total synthesis and analogues | [163][164] |
| Stellettapeptins A (54) and B (55) | Cyclic undecadepsi-peptides | Microorganisms symbiosis Stelletta sp. | EC50: infection of human T-lymphoblastoid cells by HIV-1RF (23 and 27 nM, respectively) | - | [165] |
| Stylissamide G (56) | Cyclic heptapeptide | Stylissa caribica | MIC: M. audouinii, T. mentagrophytes, C. albicans (6 μg/mL) | Total Synthesis | [166] |
| Theonegramide (57) | Bicyclic glycododecapeptide | Bacteria symbiosis Theonella swinhoei | C. albicans (10 µg/disk) |
- | [167] |
| Theonellamide G (58) | Bicyclic glyco-depsipeptide | Bacteria symbiosis Theonella swinhoei | IC50: Wild and amphotericin B-resistant strains of C. albicans (2.0–4.49 μM), amphotericin-B (1.48 μM) | Semi-synthesis | [121][168] |
| Theonellapeptolide congeners 1 (59) and 2 (60) | Cyclic trideca-depsipeptides | Theonella sp. | MIC: S. aureus (8.0–16 µg/mL), M. luteus (8.0 µg/mL), B. subtilis (8.0–16 µg/mL), M. smegmatis (16–66 µg/mL), T. mentagrophytes (4.0–8.0 µg/mL), A. niger (8.0–66 µg/mL) | Total synthesis and analogues | [169][170] |
| Theopapuamide A-C (61–63) | Cyclic undeca-depsipeptides | Bacteria symbiosis Theonella swinhoei and Siliquarias-pongia mirabilis | Wild type and amphotericin B-resistant strains of C. albicans (1–5 µg/disk); in vitro HIV-1 infectivity assay IC50 = 0.8 μg/mL |
- | [123] |
2.2. Bacteria-Produced Cyclic Peptides
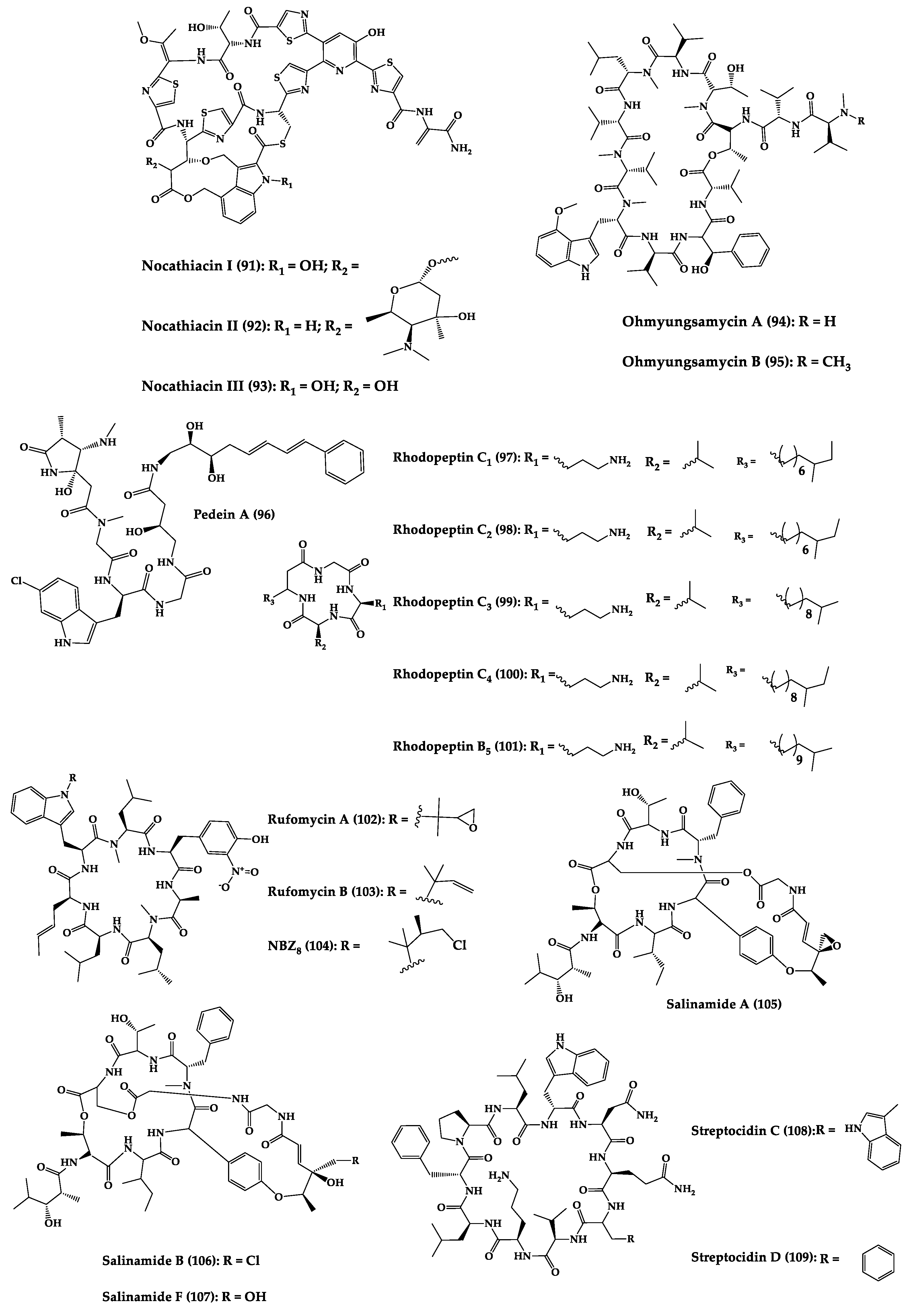
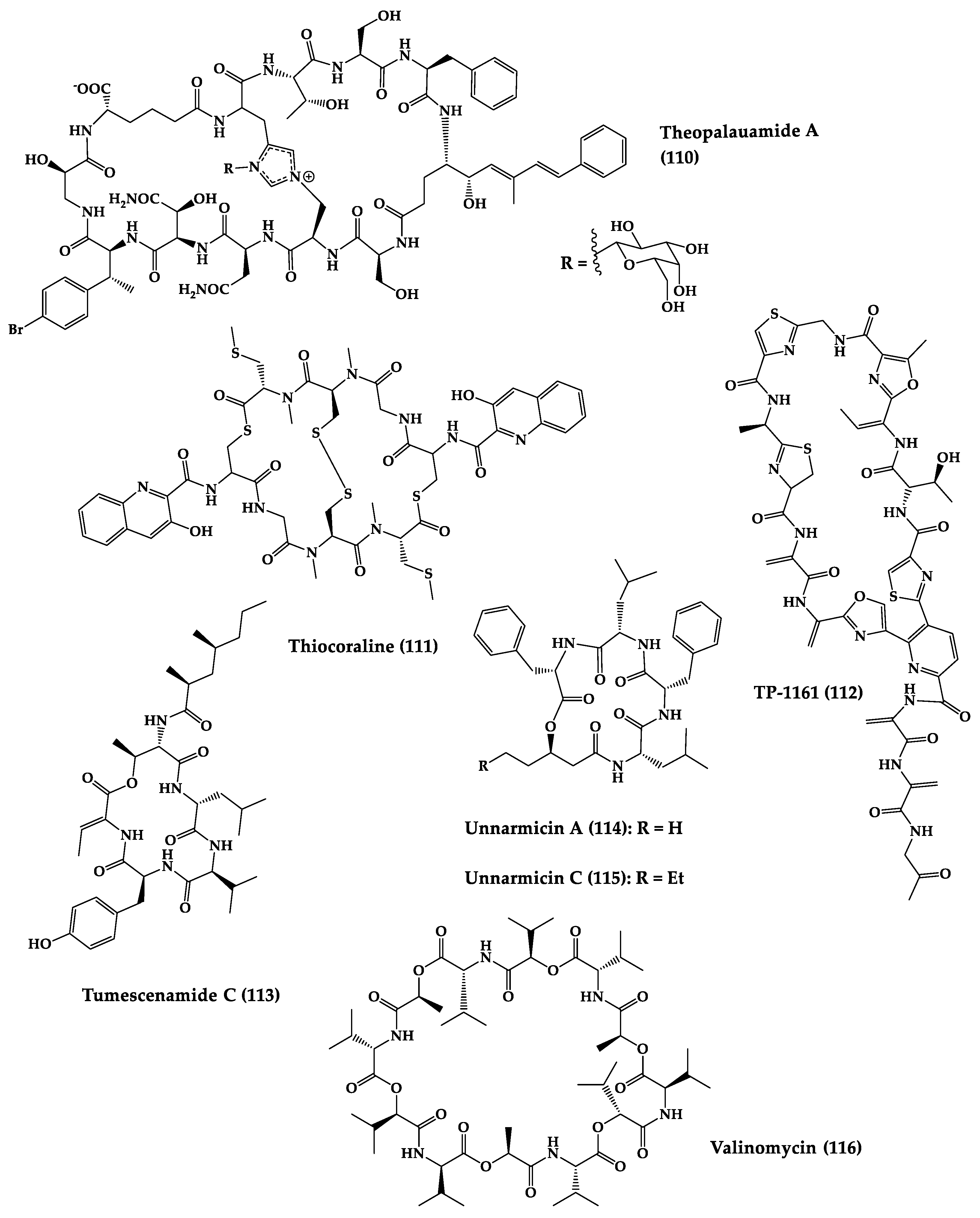
This entry is adapted from the peer-reviewed paper 10.3390/md20060397
References
- Loretz, B.; Oh, Y.-K.; Hudson, S.; Gu, Z.; Lehr, C.-M. Drug delivery for fighting infectious diseases: A global perspective. Drug Deliv. Transl. Res. 2021, 11, 1316–1322.
- WHO. Antimicrobial Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 12 April 2021).
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. Available online: https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(21)02724-0/fulltext (accessed on 12 April 2021).
- Livermore, D.M.; British Society for Antimicrobial Chemotherapy Working Party on The Urgent Need: Regenerating Antibacterial Drug Discovery and Development; Blaser, M.; Carrs, O.; Cassell, G.; Fishman, N.; Guidos, R.; Levy, S.; Powers, J.; Norrby, R.; et al. Discovery research: The scientific challenge of finding new antibiotics. J. Antimicrob. Chemother. 2011, 66, 1941–1944.
- Amyes, S.G. Magic Bullets, Lost Horizons: The Rise and Fall of Antibiotics, 1st ed.; CRC Press: London, UK, 2001; p. 272.
- Walsh, C.T.; Wencewicz, T.A. Prospects for new antibiotics: A molecule-centered perspective. J. Antibiot. 2014, 67, 7–22.
- Sun, C.; Hunt, D.K.; Clark, R.B.; Lofland, D.; O’Brien, W.J.; Plamondon, L.; Xiao, X.-Y. Synthesis and antibacterial activity of pentacyclines: A novel class of tetracycline analogs. J. Med. Chem. 2011, 54, 3704–3731.
- Campoy, S.; Adrio, J.L. Antifungals. Biochem. Pharmacol. 2017, 133, 86–96.
- Thompson, G.R.; Cadena, J.; Patterson, T.F. Overview of antifungal agents. Clin. Chest Med. 2009, 30, 203–215.
- Pfaller, M.A.; Diekema, D.J. Epidemiology of invasive candidiasis: A persistent public health problem. Clin. Microbiol. Rev. 2007, 20, 133–163.
- Caston-Osorio, J.; Rivero, A.; Torre-Cisneros, J. Epidemiology of invasive fungal infection. Int. J. Antimicrob. Agents 2008, 32, S103–S109.
- Sobel, J.D.; Nyirjesy, P. Oteseconazole: An advance in treatment of recurrent vulvovaginal candidiasis. Future Microbiol. 2021, 16, 1453–1461.
- De Clercq, E. Recent highlights in the development of new antiviral drugs. Curr. Opin. Microbiol. 2005, 8, 552–560.
- Woolhouse, M.; Scott, F.; Hudson, Z.; Howey, R.; Chase-Topping, M. Human viruses: Discovery and emergence. Philos. Trans. R. Soc. B 2012, 367, 2864–2871.
- De Clercq, E.; Li, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747.
- Wu, J.T.; Leung, K.; Leung, G.M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: A modelling study. Lancet 2020, 395, 689–697.
- Nash, T.E. Parasitic Diseases that Cause Seizures: Parasitic Diseases that Cause Seizures. Epilepsy Curr. 2014, 14, 29–34.
- Renslo, A.R.; McKerrow, J.H. Drug discovery and development for neglected parasitic diseases. Nat. Chem. Biol. 2006, 2, 701–710.
- Pozio, E. World distribution of Trichinella spp. infections in animals and humans. Vet. Parasitol. 2007, 149, 3–21.
- Garcia, H.H.; Gonzalez, A.E.; Gilman, R.H. Diagnosis, treatment and control of Taenia solium cysticercosis. Curr. Opin. Infect. Dis. 2003, 16, 411–419.
- Pink, R.; Hudson, A.; Mouriès, M.-A.; Bendig, M. Opportunities and challenges in antiparasitic drug discovery. Nat. Rev. Drug Discov. 2005, 4, 727–740.
- Watkins, B.M. Drugs for the control of parasitic diseases: Current status and development. Trends Parasitol. 2003, 19, 477–478.
- Upcroft, J.A.; Dunn, L.A.; Wal, T.; Tabrizi, S.; Delgadillo-Correa, M.G.; Johnson, P.J.; Garland, S.; Siba, P.; Upcroft, P. Metronidazole resistance in Trichomonas vaginalis from highland women in Papua New Guinea. Sex Health 2009, 6, 334–338.
- Anthwal, A.; Rajesh, U.C.; Rawat, M.; Kushwaha, B.; Maikhuri, J.P.; Sharma, V.L.; Gupta, G.; Rawat, D.S. Novel metronidazole–chalcone conjugates with potential to counter drug resistance in Trichomonas vaginalis. Eur. J. Med. Chem. 2014, 79, 89–94.
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318.
- Shrestha, P.; Cooper, B.S.; Coast, J.; Oppong, R.; Thuy, N.D.T.; Phodha, T.; Celhay, O.; Guerin, P.J.; Wertheim, H.; Lubell, Y. Enumerating the economic cost of antimicrobial resistance per antibiotic consumed to inform the evaluation of interventions affecting their use. Antimicrob. Resist. Infect. Control. 2018, 7, 1–9.
- Groseclose, S.L.; Buckeridge, D.L. Public health surveillance systems: Recent advances in their use and evaluation. Annu. Rev. Public Health 2017, 38, 57–79.
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66.
- Laxminarayan, R.; Matsoso, P.; Pant, S.; Brower, C.; Røttingen, J.-A.; Klugman, K.; Davies, S. Access to effective antimicrobials: A worldwide challenge. Lancet 2016, 387, 168–175.
- Martens, E.; Demain, A.L. The antibiotic resistance crisis, with a focus on the United States. J. Antibiot. 2017, 70, 520–526.
- Andersson, D.I.; Hughes, D.; Kubicek-Sutherland, J.Z. Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resist. Updates 2016, 26, 43–57.
- Anjum, K.; Abbas, S.Q.; Shah, S.A.A.; Akhter, N.; Batool, S.; ul Hassan, S.S. Marine sponges as a drug treasure. Biomol. Ther. 2016, 24, 347.
- Mayer, A.; Rodríguez, A.D.; Taglialatela-Scafati, O.; Fusetani, N. Marine pharmacology in 2009–2011: Marine compounds with antibacterial, antidiabetic, antifungal, anti-inflammatory, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Mar. Drugs 2013, 11, 2510–2573.
- Cheung, R.C.F.; Ng, T.B.; Wong, J.H. Marine peptides: Bioactivities and applications. Mar. Drugs 2015, 13, 4006–4043.
- Mayer, A.M.; Rodríguez, A.D.; Berlinck, R.G.; Fusetani, N. Marine pharmacology in 2007–8: Marine compounds with antibacterial, anticoagulant, antifungal, anti-inflammatory, antimalarial, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous system, and other miscellaneous mechanisms of action. Comp. Biochem. Physiol. C 2011, 153, 191–222.
- Aneiros, A.; Garateix, A. Bioactive peptides from marine sources: Pharmacological properties and isolation procedures. J. Chromatogr. B 2004, 803, 41–53.
- Donia, M.; Hamann, M.T. Marine natural products and their potential applications as anti-infective agents. Lancet Infect. Dis. 2003, 3, 338–348.
- Koslow, J.A. The silent deep: The discovery, ecology, and conservation of the deep sea. Oceanography 2007, 23, 228.
- Russo, P.; Del Bufalo, A.; Fini, M. Deep sea as a source of novel-anticancer drugs: Update on discovery and preclinical/clinical evaluation in a systems medicine perspective. EXCLI J. 2015, 14, 228.
- Xu, L.; Meng, W.; Cao, C.; Wang, J.; Shan, W.; Wang, Q. Antibacterial and antifungal compounds from marine fungi. Mar. Drugs 2015, 13, 3479–3513.
- Alves, A.; Sousa, E.; Kijjoa, A.; Pinto, M. Marine-derived compounds with potential use as cosmeceuticals and nutricosmetics. Molecules 2020, 25, 2536.
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine sponge derived natural products between 2001 and 2010: Trends and opportunities for discovery of bioactives. Mar. Drugs 2014, 12, 4539–4577.
- Bhatnagar, I.; Kim, S.-K. Immense essence of excellence: Marine microbial bioactive compounds. Mar. Drugs 2010, 8, 2673–2701.
- Hu, Y.; Chen, J.; Hu, G.; Yu, J.; Zhu, X.; Lin, Y.; Chen, S.; Yuan, J. Statistical research on the bioactivity of new marine natural products discovered during the 28 years from 1985 to 2012. Mar. Drugs 2015, 13, 202–221.
- Winder, P.L.; Pomponi, S.A.; Wright, A.E. Natural Products from the Lithistida: A Review of the Literature since 2000. Mar. Drugs 2011, 9, 2643–2682.
- Duray, H.; Hatfill, J.; Pellis, R. Venom peptides as human pharmaceuticals. Sci. Med. 1997, 4, 6–15.
- Wang, X.; Gong, X.; Li, P.; Lai, D.; Zhou, L. Structural diversity and biological activities of cyclic depsipeptides from fungi. Molecules 2018, 23, 169.
- May Zin, W.W.; Buttachon, S.; Dethoup, T.; Fernandes, C.; Cravo, S.; Pinto, M.M.; Gales, L.; Pereira, J.A.; Silva, A.; Sekeroglu, N. New cyclotetrapeptides and a new diketopiperzine derivative from the marine sponge-associated fungus Neosartorya glabra KUFA 0702. Mar. Drugs 2016, 14, 136.
- Prompanya, C.; Fernandes, C.; Cravo, S.; Pinto, M.M.; Dethoup, T.; Silva, A.; Kijjoa, A. A new cyclic hexapeptide and a new isocoumarin derivative from the marine sponge-associated fungus Aspergillus similanensis KUFA 0013. Mar. Drugs 2015, 13, 1432–1450.
- Cooper, B.M.; Iegre, J.; O’Donovan, D.H.; Halvarsson, M.Ö.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide–drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494.
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147.
- Zompra, A.A.; Galanis, A.S.; Werbitzky, O.; Albericio, F. Manufacturing peptides as active pharmaceutical ingredients. Future Med. Chem. 2009, 1, 361–377.
- Otvos, L., Jr. Peptide-based drug design: Here and now. Methods Mol. Biol. 2008, 494, 1–8.
- Goodwin, D.; Simerska, P.; Toth, I. Peptides as therapeutics with enhanced bioactivity. Curr. Med. Chem. 2012, 19, 4451–4461.
- Ahrens, V.M.; Frank, R.; Boehnke, S.; Schütz, C.L.; Hampel, G.; Iffland, D.S.; Bings, N.H.; Hey-Hawkins, E.; Beck-Sickinger, A.G. Receptor-mediated uptake of boron-rich neuropeptide y analogues for boron neutron capture therapy. ChemMedChem 2015, 10, 164–172.
- Mason, J.M. Design and development of peptides and peptide mimetics as antagonists for therapeutic intervention. Future Med. Chem. 2010, 2, 1813–1822.
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: Incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203.
- Guharoy, M.; Chakrabarti, P. Secondary structure based analysis and classification of biological interfaces: Identification of binding motifs in protein–protein interactions. Bioinformatics 2007, 23, 1909–1918.
- DeLorbe, J.E.; Clements, J.H.; Whiddon, B.B.; Martin, S.F. Thermodynamic and structural effects of macrocyclic constraints in protein–ligand interactions. ACS Med. Chem. Lett. 2010, 1, 448–452.
- DeLorbe, J.E.; Clements, J.H.; Teresk, M.G.; Benfield, A.P.; Plake, H.R.; Millspaugh, L.E.; Martin, S.F. Thermodynamic and Structural Effects of Conformational Constraints in Protein–Ligand Interactions. Entropic Paradoxy Associated with Ligand Preorganization. J. Am. Chem. Soc. 2009, 131, 16758–16770.
- Tapeinou, A.; Matsoukas, M.T.; Simal, C.; Tselios, T. Review cyclic peptides on a merry-go-round; towards drug design. Pept. Sci. 2015, 104, 453–461.
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into protein–ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144.
- Lee, M.S.; Gardner, B.; Kahn, M.; Nakanishi, H. The three dimensional solution structure of a constrained peptidomimetic in water and in chloroform observation of solvent induced hydrophobic cluster. FEBS Lett. 1995, 359, 113–118.
- Uma, K.; Kishore, R.; Balaram, P. Stereochemical constraints in peptide design: Analysis of the influence of a disulfide bridge and an α-aminoisobutyryl residue on the conformation of a hexapeptide. Biopolymers 1993, 33, 865–871.
- Lau, D.; Guo, L.; Liu, R.; Marik, J.; Lam, K. Peptide ligands targeting integrin α3β1 in non-small cell lung cancer. Lung Cancer 2006, 52, 291–297.
- Rezai, T.; Yu, B.; Millhauser, G.L.; Jacobson, M.P.; Lokey, R.S. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J. Am. Chem. Soc. 2006, 128, 2510–2511.
- Kwon, Y.-U.; Kodadek, T. Quantitative comparison of the relative cell permeability of cyclic and linear peptides. Chem. Biol. 2007, 14, 671–677.
- Hussack, G.; Hirama, T.; Ding, W.; MacKenzie, R.; Tanha, J. Engineered single-domain antibodies with high protease resistance and thermal stability. PLoS ONE 2011, 6, e28218.
- Lindgren, M.; Hällbrink, M.; Prochiantz, A.; Langel, Ü. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000, 21, 99–103.
- Cini, E.; Bifulco, G.; Menchi, G.; Rodriquez, M.; Taddei, M. Synthesis of Enantiopure 7-substituted Azepane-2-carboxylic acids as templates for conformationally constrained Peptidomimetics. Eur. J. Org. Chem. 2012, 2012, 2133–2141.
- Grigoryan, G.; Reinke, A.W.; Keating, A.E. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature 2009, 458, 859–864.
- Grauer, A.; König, B. Peptidomimetics—A versatile route to biologically active compounds. Eur. J. Org. Chem. 2009, 2009, 5099–5111.
- Mandell, D.J.; Kortemme, T. Computer-aided design of functional protein interactions. Nat. Chem. Biol. 2009, 5, 797–807.
- Terrett, N. Drugs in middle space. MedChemComm 2013, 4, 474–475.
- Joo, S.H. Cyclic peptides as therapeutic agents and biochemical tools. Biomol. Ther. 2012, 20, 19.
- Gerwick, W.H.; Fenner, A.M. Drug discovery from marine microbes. Microb. Ecol. 2013, 65, 800–806.
- Rocha-Martin, J.; Harrington, C.; Dobson, A.D.; O’Gara, F. Emerging strategies and integrated systems microbiology technologies for biodiscovery of marine bioactive compounds. Mar. Drugs 2014, 12, 3516–3559.
- Kang, H.K.; Seo, C.H.; Park, Y. Marine peptides and their anti-infective activities. Mar. Drugs 2015, 13, 618–654.
- Bionda, N.; Stawikowski, M.; Stawikowska, R.; Cudic, M.; López-Vallejo, F.; Treitl, D.; Medina-Franco, J.; Cudic, P. Effects of cyclic lipodepsipeptide structural modulation on stability, antibacterial activity, and human cell toxicity. ChemMedChem 2012, 7, 871–882.
- Raaijmakers, J.M.; De Bruijn, I.; Nybroe, O.; Ongena, M. Natural functions of lipopeptides from Bacillus and Pseudomonas: More than surfactants and antibiotics. FEMS Microbiol. Rev. 2010, 34, 1037–1062.
- Jensen, K.J.; Brask, J. Carbohydrates in peptide and protein design. Pept. Sci. 2005, 80, 747–761.
- Taevernier, L.; Wynendaele, E.; Gevaert, B.; De Spiegeleer, B. Chemical classification of cyclic depsipeptides. Curr. Protein Pept. Sci. 2017, 18, 425–452.
- Moss, G.; Smith, P.; Tavernier, D. Glossary of class names of organic compounds and reactivity intermediates based on structure (IUPAC Recommendations 1995). Pure Appl. Chem. 1995, 67, 1307–1375.
- Dixon, H.B.F. Nomenclature and symbolism for amino acids and peptides. Pure Appl. Chem. 1984, 56, 595–624. Available online: https://febs.onlinelibrary.wiley.com/doi/10.1111/j.1432-1033.1984.tb07877.x (accessed on 12 April 2021).
- Sieber, S.A.; Marahiel, M.A. Molecular mechanisms underlying nonribosomal peptide synthesis: Approaches to new antibiotics. Chem. Rev. 2005, 105, 715–738.
- Fischbach, M.A.; Walsh, C.T. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem. Rev. 2006, 106, 3468–3496.
- Thompson, F.T.C. Biossíntese de Metabólitos Secundários. In Biotecnologia Marinha; FURG, Ed.; PPG-Mar: Rio Grande, Mexico, 2020; pp. 91–95.
- Reimer, J.M.; Haque, A.S.; Tarry, M.J.; Schmeing, T.M. Piecing together nonribosomal peptide synthesis. Curr. Opin. Struct. Biol. 2018, 49, 104–113.
- Gulick, A.M. Structural insight into the necessary conformational changes of modular nonribosomal peptide synthetases. Curr. Opin. Chem. Biol. 2016, 35, 89–96.
- Schwarzer, D.; Finking, R.; Marahiel, M.A. Nonribosomal peptides: From genes to products. Nat. Prod. Rep. 2003, 20, 275–287.
- Marahiel, M.A.; Stachelhaus, T.; Mootz, H.D. Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem. Rev. 1997, 97, 2651–2674.
- Alonzo, D.A.; Schmeing, T.M. Biosynthesis of depsipeptides, or Depsi: The peptides with varied generations. Protein Sci. 2020, 29, 2316–2347.
- Donadio, S.; Monciardini, P.; Sosio, M. Polyketide synthases and nonribosomal peptide synthetases: The emerging view from bacterial genomics. Nat. Prod. Rep. 2007, 24, 1073–1109.
- Horsman, M.E.; Hari, T.P.; Boddy, C.N. Polyketide synthase and non-ribosomal peptide synthetase thioesterase selectivity: Logic gate or a victim of fate? Nat. Prod. Rep. 2016, 33, 183–202.
- Wilson, D.J.; Shi, C.; Teitelbaum, A.M.; Gulick, A.M.; Aldrich, C.C. Characterization of AusA: A dimodular nonribosomal peptide synthetase responsible for the production of aureusimine pyrazinones. Biochemistry 2013, 52, 926–937.
- Manavalan, B.; Murugapiran, S.K.; Lee, G.; Choi, S. Molecular modeling of the reductase domain to elucidate the reaction mechanism of reduction of peptidyl thioester into its corresponding alcohol in non-ribosomal peptide synthetases. BMC Struct. Biol. 2010, 10, 1–14.
- Bloudoff, K.; Schmeing, T.M. Structural and functional aspects of the nonribosomal peptide synthetase condensation domain superfamily: Discovery, dissection and diversity. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1587–1604.
- Zhang, J.; Liu, N.; Cacho, R.A.; Gong, Z.; Liu, Z.; Qin, W.; Tang, C.; Tang, Y.; Zhou, J. Structural basis of nonribosomal peptide macrocyclization in fungi. Nat. Chem. Biol. 2016, 12, 1001–1003.
- Bloudoff, K.; Fage, C.D.; Marahiel, M.A.; Schmeing, T.M. Structural and mutational analysis of the nonribosomal peptide synthetase heterocyclization domain provides insight into catalysis. Proc. Natl. Acad. Sci. USA 2017, 114, 95–100.
- Yuwen, L.; Zhang, F.-L.; Chen, Q.-H.; Lin, S.-J.; Zhao, Y.-L.; Li, Z.-Y. The role of aromatic L-amino acid decarboxylase in bacillamide C biosynthesis by Bacillus atrophaeus C89. Sci. Rep. 2013, 3, 1–10.
- Chang, C.-Y.; Lohman, J.R.; Huang, T.; Michalska, K.; Bigelow, L.; Rudolf, J.D.; Jedrzejczak, R.; Yan, X.; Ma, M.; Babnigg, G. Structural insights into the free-standing condensation enzyme SgcC5 catalyzing ester-bond formation in the biosynthesis of the enediyne antitumor antibiotic C-1027. Biochemistry 2018, 57, 3278–3288.
- Anand, T.P.; Chellaram, C.; Kuberan, G.; Archana, H. Bioactive peptides from marine sources-a review. Indian J. Innov. Dev. 2012, 1, 61–64.
- Bewley, C.A.; Faulkner, D.J. Lithistid sponges: Star performers or hosts to the stars. Angew. Chem. Int. Ed. 1998, 37, 2162–2178.
- Schmidt, E.; Obraztsova, A.; Davidson, S.; Faulkner, D.; Haygood, M. Identification of the antifungal peptide-containing symbiont of the marine sponge Theonella swinhoei as a novel δ-proteobacterium,“Candidatus Entotheonella palauensis”. Mar. Biol. 2000, 136, 969–977.
- Amelia, T.S.M.; Suaberon, F.A.C.; Vad, J.; Fahmi, A.D.M.; Saludes, J.P.; Bhubalan, K. Recent Advances of Marine Sponge-Associated Microorganisms as a Source of Commercially Viable Natural Products. Mar. Biotechnol. 2022, 1–21.
- Liu, L.; Zheng, Y.-Y.; Shao, C.-L.; Wang, C.-Y. Metabolites from marine invertebrates and their symbiotic microorganisms: Molecular diversity discovery, mining, and application. Mar. Life Sci. Technol. 2019, 1, 60–94.
- Kobayashi, M.; Tanaka, J.-I.; Katori, T.; Matsuura, M.; Yamashita, M.; Kitagawa, I. Marine natural products. XXII: The absolute stereostructure of swinholide A, a potent cytotoxic dimeric macrolide from the Okinawan marine sponge Theonella swinhoei. Chem. Pharm. Bull. 1990, 38, 2409–2418.
- Li, H.-Y.; Matsunaga, S.; Fusetani, N. Halicylindramides A-C, antifungal and cytotoxic depsipeptides from the marine sponge Halichondria cylindrata. J. Med. Chem. 1995, 38, 338–343.
- Zampella, A.; Sepe, V.; Luciano, P.; Bellotta, F.; Monti, M.C.; D’Auria, M.V.; Jepsen, T.; Petek, S.; Adeline, M.-T.; Laprévôte, O. Homophymine A, an anti-HIV cyclodepsipeptide from the sponge Homophymia sp. J. Org. Chem. 2008, 73, 5319–5327.
- Zabriskie, T.M.; Klocke, J.A.; Ireland, C.M.; Marcus, A.H.; Molinski, T.F.; Faulkner, D.J.; Xu, C.; Clardy, J. Jaspamide, a modified peptide from a Jaspis sponge, with insecticidal and antifungal activity. J. Am. Chem. Soc. 1986, 108, 3123–3124.
- Scott, V.; Boehme, R.; Matthews, T. New class of antifungal agents: Jasplakinolide, a cyclodepsipeptide from the marine sponge, Jaspis species. Antimicrob. Agents Chemother. 1988, 32, 1154–1157.
- Fusetani, N.; Matsunaga, S. Bioactive sponge peptides. Chem. Rev. 1993, 93, 1793–1806.
- Plaza, A.; Bifulco, G.; Masullo, M.; Lloyd, J.R.; Keffer, J.L.; Colin, P.L.; Hooper, J.N.; Bell, L.J.; Bewley, C.A. Mutremdamide A and koshikamides C–H, peptide inhibitors of HIV-1 entry from different Theonella species. J. Org. Chem. 2010, 75, 4344–4355.
- Rashid, M.A.; Gustafson, K.R.; Cartner, L.K.; Shigematsu, N.; Pannell, L.K.; Boyd, M.R. Microspinosamide, a New HIV-Inhibitory Cyclic Depsipeptide from the Marine Sponge Sidonops microspinosa. J. Nat. Prod. 2001, 64, 117–121.
- Plaza, A.; Gustchina, E.; Baker, H.L.; Kelly, M.; Bewley, C.A. Mirabamides A–D, depsipeptides from the sponge Siliquariaspongia mirabilis that inhibit HIV-1 fusion. J. Nat. Prod. 2007, 70, 1753–1760.
- Lu, Z.; Van Wagoner, R.M.; Harper, M.K.; Baker, H.L.; Hooper, J.N.; Bewley, C.A.; Ireland, C.M. Mirabamides E–H, HIV-inhibitory depsipeptides from the sponge Stelletta clavosa. J. Nat. Prod. 2011, 74, 185–193.
- Gulakowski, R.J.; McMahon, J.B.; Staley, P.G.; Moran, R.A.; Boyd, M.R. A semiautomated multiparameter approach for anti-HIV drug screening. J. Virol. Methods 1991, 33, 87–100.
- Oku, N.; Gustafson, K.R.; Cartner, L.K.; Wilson, J.A.; Shigematsu, N.; Hess, S.; Pannell, L.K.; Boyd, M.R.; McMahon, J.B. Neamphamide A, a new HIV-inhibitory depsipeptide from the Papua New Guinea marine sponge Neamphius huxleyi. J. Nat. Prod. 2004, 67, 1407–1411.
- Yamano, Y.; Arai, M.; Kobayashi, M. Neamphamide B, new cyclic depsipeptide, as an anti-dormant mycobacterial substance from a Japanese marine sponge of Neamphius sp. Bioorg. Med. Chem. Lett. 2012, 22, 4877–4881.
- Ford, P.W.; Gustafson, K.R.; McKee, T.C.; Shigematsu, N.; Maurizi, L.K.; Pannell, L.K.; Williams, D.E.; Dilip de Silva, E.; Lassota, P.; Allen, T.M. Papuamides A–D, HIV-Inhibitory and Cytotoxic Depsipeptides from the Sponges Theonella mirabilis and Theonella swinhoei Collected in Papua New Guinea. J. Am. Chem. Soc. 1999, 121, 5899–5909.
- Youssef, D.T.; Shaala, L.A.; Mohamed, G.A.; Badr, J.M.; Bamanie, F.H.; Ibrahim, S.R. Theonellamide G, a potent antifungal and cytotoxic bicyclic glycopeptide from the Red Sea marine sponge Theonella swinhoei. Mar. Drugs 2014, 12, 1911–1923.
- Ratnayake, A.S.; Bugni, T.S.; Feng, X.; Harper, M.K.; Skalicky, J.J.; Mohammed, K.A.; Andjelic, C.D.; Barrows, L.R.; Ireland, C.M. Theopapuamide, a cyclic depsipeptide from a Papua New Guinea lithistid sponge Theonella swinhoei. J. Nat. Prod. 2006, 69, 1582–1586.
- Plaza, A.; Bifulco, G.; Keffer, J.L.; Lloyd, J.R.; Baker, H.L.; Bewley, C.A. Celebesides A–C and theopapuamides B–D, depsipeptides from an Indonesian sponge that inhibit HIV-1 entry. J. Org. Chem. 2009, 74, 504–512.
- Bewley, C.A.; He, H.; Williams, D.H.; Faulkner, D.J. Aciculitins A–C: Cytotoxic and antifungal cyclic peptides from the lithistid sponge Aciculites orientalis. J. Am. Chem. Soc. 1996, 118, 4314–4321.
- Ng-Choi, I.; Oliveras, À.; Feliu, L.; Planas, M. Solid-phase synthesis of biaryl cyclic peptides containing a histidine-phenylalanine linkage. Int. J. Pept. Res. Ther. 2020, 26, 695–707.
- Zampella, A.; D’Auria, M.V.; Paloma, L.G.; Casapullo, A.; Minale, L.; Debitus, C.; Henin, Y. Callipeltin A, an anti-HIV cyclic depsipeptide from the New Caledonian Lithistida sponge Callipelta sp. J. Am. Chem. Soc. 1996, 118, 6202–6209.
- Liang, B.; Carroll, P.J.; Joullié, M.M. Progress toward the total synthesis of callipeltin A (I): Asymmetric synthesis of (3S, 4 R)-3,4-dimethylglutamine. Org. Lett. 2000, 2, 4157–4160.
- Hansen, D.B.; Wan, X.; Carroll, P.J.; Joullié, M.M. Stereoselective synthesis of four stereoisomers of β-methoxytyrosine, a component of callipeltin A. J. Org. Chem. 2005, 70, 3120–3126.
- Kikuchi, M.; Konno, H. Total synthesis of callipeltin B and M, peptidyl marine natural products. Org. Lett. 2014, 16, 4324–4327.
- Zampella, A.; D’Auria, M.V. Stereoselective synthesis of (2R,3R,4R)-3-hydroxy-2,4,6-trimethylheptanoic acid and determination of the absolute stereochemistry of the natural product from callipeltin A. Tetrahedron Asymmetry 2002, 13, 1237–1239.
- Daletos, G.; Kalscheuer, R.; Koliwer-Brandl, H.; Hartmann, R.; De Voogd, N.J.; Wray, V.; Lin, W.; Proksch, P. Callyaerins from the marine sponge Callyspongia aerizusa: Cyclic peptides with antitubercular activity. J. Nat. Prod. 2015, 78, 1910–1925.
- Zhang, S.; De Leon Rodriguez, L.M.; Leung, I.K.; Cook, G.M.; Harris, P.W.; Brimble, M.A. Total Synthesis and Conformational Study of Callyaerin A: Anti-Tubercular Cyclic Peptide Bearing a Rare Rigidifying (Z)-2,3-Diaminoacrylamide Moiety. Angew. Chem. Int. Ed. 2018, 57, 3631–3635.
- Clark, D.P.; Carroll, J.; Naylor, S.; Crews, P. An antifungal cyclodepsipeptide, cyclolithistide A, from the sponge Theonella swinhoei. J. Org. Chem. 1998, 63, 8757–8764.
- Grieco, P.A.; Perez-Medrano, A. Total synthesis of the mixed peptide-polypropionate based cyclodepsipeptide (+)-geodiamolide B. Tetrahedron Lett. 1988, 29, 4225–4228.
- Chan, W.R.; Tinto, W.F.; Manchand, P.S.; Todaro, L.J. Stereostructures of geodiamolides A and B, novel cyclodepsipeptides from the marine sponge Geodia sp. J. Org. Chem. 1987, 52, 3091–3093.
- White, J.D.; Amedio, J.C., Jr. Total synthesis of geodiamolide A, a novel cyclodepsipeptide of marine origin. J. Org. Chem. 1989, 54, 736–738.
- Amagata, T.; Morinaka, B.I.; Amagata, A.; Tenney, K.; Valeriote, F.A.; Lobkovsky, E.; Clardy, J.; Crews, P. A chemical study of cyclic depsipeptides produced by a sponge-derived fungus. J. Nat. Prod. 2006, 69, 1560–1565.
- Yeo, S.-H.; Seo, H.-J.; Lim, D.-Y. Synthesis of halicylindramide a mimetics containing lactone isosteres. Bull. Korean Chem. Soc. 2011, 32, 2916–2920.
- Seo, H.; Lim, D. Total synthesis of Halicylindramide A. J. Org. Chem. 2009, 74, 906–909.
- Bellotta, F.; D’Auria, M.V.; Sepe, V.; Zampella, A. Synthetic studies on homophymine A: Stereoselective synthesis of (2R,3R,4R,6R)-3-hydroxy-2,4,6-trimethyloctanoic acid. Tetrahedron 2009, 65, 3659–3663.
- Ohtaka, J.; Hamajima, A.; Nemoto, T.; Hamada, Y. Efficient diastereoselective synthesis of (2R, 3R, 4R)-2-amino-3-hydroxy-4, 5-dimethylhexanoic acid, the lactone linkage unit of homophymine A. Chem. Pharm. Bull. 2013, 61, 245–250.
- Kobayashi, J.I.; Tsuda, M.; Nakamura, T.; Mikami, Y.; Shigemori, H. Hymenamides A and B, new proline-rich cyclic heptapeptides from the Okinawan marine sponge Hymeniacidon sp. Tetrahedron 1993, 49, 2391–2402.
- Shiki, Y.; Onai, M.; Sugiyama, D.; Osada, S.; Fujita, I.; Kodama, H. Synthesis and biological activities of cyclic peptide, hymenamide analogs. In Peptides for Youth; Springer: New York, NY, USA, 2009; pp. 323–324.
- Tsuda, M.; Shigemori, H.; Mikami, Y.; Kobayashi, J.i. Hymenamides C-E, new cyclic heptapeptides with two proline residues from the okinawan marine sponge hymeniacidon sp. Tetrahedron 1993, 49, 6785–6796.
- Chu, K.S.; Negrete, G.R.; Konopelski, J.P. Asymmetric total synthesis of (+)-jasplakinolide. J. Org. Chem. 1991, 56, 5196–5202.
- Grieco, P.A.; Hon, Y.S.; Perez-Medrano, A. Convergent, enantiospecific total synthesis of the novel cyclodepsipeptide (+)-jasplakinolide (jaspamide). J. Am. Chem. Soc. 1988, 110, 1630–1631.
- Ghosh, A.K.; Moon, D.K. Enantioselective total synthesis of (+)-jasplakinolide. Org. Lett. 2007, 9, 2425–2427.
- Davis, R.A.; Mangalindan, G.C.; Bojo, Z.P.; Antemano, R.R.; Rodriguez, N.O.; Concepcion, G.P.; Samson, S.C.; de Guzman, D.; Cruz, L.J.; Tasdemir, D. Microcionamides A and B, bioactive peptides from the Philippine sponge Clathria (Thalysias) abietina. J. Org. Chem. 2004, 69, 4170–4176.
- Bewley, C.A.; Debitus, C.; Faulkner, D.J. Microsclerodermins A and B. Antifungal cyclic peptides from the lithistid sponge Microscleroderma sp. J. Am. Chem. Soc. 1994, 116, 7631–7636.
- Schmidt, E.W.; Faulkner, D.J. Microsclerodermins C-E, antifungal cyclic peptides from the lithistid marine sponges Theonella sp. and Microscleroderma sp. Tetrahedron 1998, 54, 3043–3056.
- Qureshi, A.; Colin, P.L.; Faulkner, D.J. Microsclerodermins F–I, Antitumor and Antifungal Cyclic Peptides from the Lithistid Sponge Microscleroderma sp. Tetrahedron 2000, 56, 3679–3685.
- Zhang, X.; Jacob, M.R.; Rao, R.R.; Wang, Y.-H.; Agarwal, A.K.; Newman, D.J.; Khan, I.A.; Clark, A.M.; Li, X.-C. Antifungal cyclic peptides from the marine sponge Microscleroderma herdmani. Med. Chem. Res. 2012, 2, 7.
- Zhu, J.; Ma, D. Total synthesis of microsclerodermin E. Angew. Chem. Int. Ed. 2003, 42, 5348–5351.
- Santhakumar, G.; Payne, R.J. Studies toward the Total Synthesis and Stereochemical Assignment of Microspinosamide. Aust. J. Chem. 2016, 68, 1885–1889.
- Ramamoorthy, G.; Acevedo, C.M.; Alvira, E.; Lipton, M.A. Synthesis and spectroscopic correlation of the diastereoisomers of 2,3-dihydroxy-2,6,8-trimethyldeca-(4Z,6E)-dienoic acid: Implications for the structures of papuamides A–D and mirabamides A–D. Tetrahedron Asymmetry 2008, 19, 2546–2554.
- Okada, Y.; Matsunaga, S.; van Soest, R.W.; Fusetani, N. Nagahamide A, an antibacterial depsipeptide from the marine sponge Theonella swinhoei. Org. Lett. 2002, 4, 3039–3042.
- Mohapatra, D.K.; Chaudhuri, S.R.; Sahoo, G.; Gurjar, M.K. Stereoselective synthesis of the polyketide chain of nagahamide A. Tetrahedron Asymmetry 2006, 17, 2609–2616.
- D’Auria, M.V.; Paloma, L.G.; Minale, L.; Zampella, A.; Debitus, C.; Perez, J. Neosiphoniamolide A, a novel cyclodepsipeptide, with antifungal activity from the marine sponge Neosiphonia superstes. J. Nat. Prod. 1995, 58, 121–123.
- Makino, K.; Nagata, E.; Hamada, Y. Synthesis of tripeptide hydrolysate from papuamide A: Determination of absolute stereostructure of β-methoxytyrosine. Tetrahedron Lett. 2005, 46, 6827–6830.
- Okamoto, N.; Hara, O.; Makino, K.; Hamada, Y. Diastereoselective synthesis of all stereoisomers of β-methoxytyrosine, a component of papuamides. J. Org. Chem. 2002, 67, 9210–9215.
- Xie, W.; Ding, D.; Zi, W.; Li, G.; Ma, D. Total synthesis and structure assignment of papuamide B, a potent marine cyclodepsipeptide with anti-HIV properties. Angew. Chem. 2008, 120, 2886–2890.
- Makino, K.; Nagata, E.; Hamada, Y. Practical synthesis of (2S,3R)-3hydroxy-3-methylproline, a constituent of papuamides, using a diastereoselective tandem Michael-aldol reaction. Tetrahedron Lett. 2005, 46, 8159–8162.
- Gulavita, N.K.; Gunasekera, S.P.; Pomponi, S.A.; Robinson, E.V. Polydiscamide A: A new bioactive depsipeptide from the marine sponge Discodermia sp. J. Org. Chem. 1992, 57, 1767–1772.
- Santhakumar, G.; Payne, R.J. Total synthesis of polydiscamides B, C, and D via a convergent native chemical ligation–oxidation strategy. Org. Lett. 2014, 16, 4500–4503.
- Shin, H.J.; Rashid, M.A.; Cartner, L.K.; Bokesch, H.R.; Wilson, J.A.; McMahon, J.B.; Gustafson, K.R. Stellettapeptins A and B, HIV-inhibitory cyclic depsipeptides from the marine sponge Stelletta sp. Tetrahedron Lett. 2015, 56, 4215–4219.
- Dahiya, R.; Singh, S.; Sharma, A.; Chennupati, S.V.; Maharaj, S. First total synthesis and biological screening of a proline-rich cyclopeptide from a Caribbean marine sponge. Mar. Drugs 2016, 14, 228.
- Bewley, C.A.; Faulkner, D.J. Theonegramide, an antifungal glycopeptide from the Philippine lithistid sponge Theonella swinhoei. J. Org. Chem. 1994, 59, 4849–4852.
- Tohdo, K.; Hamada, Y.; Shioiri, T. Theonellamide F synthetic studies. Stereoselective synthesis of (3S, 4S, 5E, 7E)-3-amino-8-(4-bromophenyl)-4-hydroxy-6-methyl-5, 7-octadienoic acid (aboa). Tetrahedron Lett. 1992, 33, 2031–2034.
- Tsuda, M.; Shimbo, K.; Kubota, T.; Mikami, Y.; Kobayashi, J.i. Two theonellapeptolide congeners from marine sponge Theonella sp. Tetrahedron 1999, 55, 10305–10314.
- Kuranaga, T.; Enomoto, A.; Tan, H.; Fujita, K.; Wakimoto, T. Total synthesis of theonellapeptolide Id. Org. Lett. 2017, 19, 1366–1369.
- Stincone, P.; Brandelli, A. Marine bacteria as source of antimicrobial compounds. Crit. Rev. Biotechnol. 2020, 40, 306–319.