Enoxacin is a second-generation quinolone with promising anticancer activity. In contrast to other members of the quinolone group, it exhibits an extraordinary cytotoxic mechanism of action. Enoxacin enhances RNA interference and promotes microRNA processing, as well as the production of free radicals. Interestingly, apart from its proapoptotic, cell cycle arresting and cytostatic effects, enoxacin manifests a limitation of cancer invasiveness. The underlying mechanisms are the competitive inhibition of vacuolar H+-ATPase subunits and c-Jun N-terminal kinase signaling pathway suppression. The mentioned mechanisms seem to contribute to a safer, more selective and more effective anticancer therapy.
- enoxacin
- miRNA
- RISC
- SMER
1. The miRNA Biogenesis
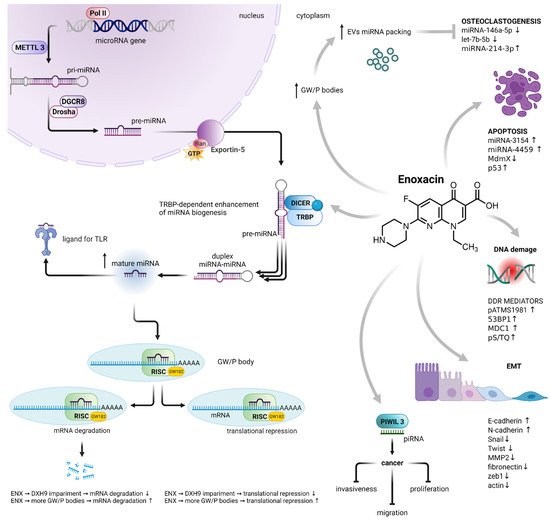
2. The Effect on Cancer Cells
2.1. TRBP-Dependent Cytotoxicity
2.2. PIWIL-3-Dependent Cytotoxicity
2.3. Other Consequences of Enoxacin-Mediated miRNA Dysregulation
3. The Effects on Non-Cancer Cells
| miRNA | Conc. [µM] | Effect | Expression Change | Cell Line | Ref. |
|---|---|---|---|---|---|
| [↑/↓] | Change-Fold | ||||
| Cancer cells | |||||
| let-7b-5p, miR-146a-5p, miR-689 | 50 | ↓ | 0.5–1 | 4T1 (miRNA from EV), | [35] |
| miR-100 | 124 | ↓ | 0.5–1 | primary ESFT spheres | [23] |
| miR-141, miR-191 | 124 | ↓ | 1.5–2 | DU145, LNcap, | [24] |
| miR-21-5p, miR-30a-3p, miR-30a-5p, miR-100-5p, miR-204-5p, miR-221-3p | 124 | ↑ | <1.5 | Cal62, STA-ET-8.2, TPC1 | [25] |
| Let-7f, miR-26a, | 124 | ↑ | <1.5 | A673, SW1736 | [23] |
| miR-21 | 100 | ↑ | 1.5–2 | MCF7 | [27] |
| miR-16, miR-18a*, miR-21, miR-26a, miR-29b, miR-29c, miR-31, miR-193a, | 124 | ↑ | 1.5–2 | HCT-116 | [22] |
| let-7f, miR-26a, miR-99a, miR-100, miR-143, miR-145, | 124 | ↑ | 1.5–2 | A673, STA-ET-8.2, TC252, primary ESFT spheres | [23] |
| miR-21-5p, miR-30a-3p, miR-100-5p, miR-146b-5p, miR-221-3p, | 124 | ↑ | 1.5–2 | Cal62, SW1736, TPC1 | [25] |
| miR-17 *, miR29b, miR-132, miR-146a, miR-191 miR-449a, | 124 | ↑ | 1.5–2 | DU145 LNcap, | [24] |
| miR-214-3p | 50 | ↑ | 2–2.5 | 4T1 (cytosolic miRNA), | [35] |
| miR-145 | 100 | ↑ | 2–2.5 | MCF7 | [27] |
| miR-7, miR-16, miR-18a*, miR-29c, miR-101, miR-128, miR-181a, miR-212 | 124 | ↑ | 2–2.5 | HCT-116, RKO | [22] |
| miR-100-5p, miR-146b-5p | 124 | ↑ | 2–2.5 | SW1736, TPC1 | [25] |
| miR-34a, miR-449a | 124 | ↑ | 2–2.5 | DU145, LNcap | [24] |
| let-7f, miR-99a, miR-100, miR-145 | 124 | ↑ | 2–2.5 | A673, STA-ET-8.2, TC252, primary ESFT spheres | [23] |
| miR-7, miR-26a, miR-29b, miR-30a, miR-101, miR-122, miR-125a, miR-125b, miR-126, miR-128, miR-143, miR-181b, miR-205 | 124 | ↑ | 2.5–3 | HCT-116, RKO | [22] |
| miR-100, miR-145 | 124 | ↑ | 2.5–3 | A673, TC252 | [23] |
| miR-29b | 124 | ↑ | 2.5–3 | LNcap | [24] |
| let-7a, let-7b, miR-30a, miR-31, miR-126, miR-181b, miR-193a, miR-193b, | 124 | ↑ | 3–3.5 | HCT-116, RKO | [22] |
| let-7f, miR-143, miR-181a, | 124 | ↑ | 3–3.5 | A673, STA-ET-8.2, primary ESFT spheres | [23] |
| miR-181a, miR-193b | 124 | ↑ | 3.5–4 | HCT-116 | [22] |
| let-7b, miR-143, miR-205 | 124 | ↑ | 4–4.5 | HCT-116, RKO | [22] |
| miR-143 | 124 | ↑ | 4–4.5 | TC252 | [23] |
| miR-125a | 124 | ↑ | ca. 5 | HCT-116 | [22] |
| miR-214-3p | 50 | ↑ | ca. 22 | 4T1 (miRNA from EV) | [35] |
| Non-cancer cells | |||||
| miR-128-1 | 60 | ↓ | 0.5–1 | dnTGFβRII T cells | [51] |
| let-7i, miR-128 | 50 | ↓ | 1.5–2 | HEK293 | [21] |
| let-7b, miR-23a, miR-30e, miR-96, miR-99a, miR-125a, miR-146, miR-190, miR-199a*, | 50 | ↑ | 1.5–2 | HEK293 | [21] |
| miR-124a, miR-139, miR-152, miR-199b | 50 | ↑ | 2–2.5 | HEK293 | [21] |
| miR-29b-1, miR-145a-5p, miR-326-3p | 60 | ↑ | 2–2.5 | dnTGFβRII T cells | [51] |
| miR-181a | 60 | ↑ | 2.5–3 | dnTGFβRII T cells | [51] |
| miR-346-5 | 60 | ↑ | 3–3.5 | dnTGFβRII T cells | [51] |
| miRNA | Dose | Effect | Expression Change: | Tissue | Ref. |
|---|---|---|---|---|---|
| [↑/↓] | Change-Fold | ||||
| miR-124 | 10 mg/kg 25 mg/kg |
↑ | ca. 4. ca. 6 |
rat frontal cortex | [52] |
| let-7a, miR-125a-5p | 10 mg/kg 25 mg/kg |
↑ | ca. 11. ca. 20 |
||
| miR-132 | 10 mg/kg 25 mg/kg |
↑ | ca. 19 (for both doses) |
||
| miR-30a-5p, miR-146b-5 | 15 mg/kg | ↑ | 1.5–2 | human orthotopic thyroid tumor from Cal62-luc mouse | [25] |
| mIR-100-5p, miR-30-3p, miR-204-5 | 15 mg/kg | ↑ | 2–2.5 | human orthotopic thyroid tumor from Cal62-luc mouse | [25] |
| miR-16, miR-18a*, miR-21, miR-26a, miR-29b, miR-29c, miR-31, miR-101, miR-193a | 10 mg/kg | ↑ | 1.5–2 | tumor from HCT-116 mouse xenograft | [22] |
| miR-16, miR-29c, miR-31, miR-101, miR-181a | 10 mg/kg | ↑ | 1.5–2 | tumor from RKO mouse xenograft | [22] |
| miR-128, miR-212 | 10 mg/kg | ↑ | 2–2.5 | tumor from HCT-116 mouse xenograft | [22] |
| miR-18a*, miR-21, miR-26a, miR-29b, miR-30a, miR-128 | 10 mg/kg | ↑ | 2–2.5 | tumor from RKO mouse xenograft | [22] |
| let-7b, miR-7, miR-143, miR-181b, miR-125b | 10 mg/kg | ↑ | 2.5–3 | tumor from HCT-116 mouse xenograft | [22] |
| let-7a, miR-7, miR-122, miR-125a, miR-125b, miR-126, miR-181b, miR-193a, miR-193b, miR-205, miR-212 | 10 mg/kg | ↑ | 2.5–3 | tumor from RKO mouse xenograft | [22] |
| let-7a, miR-30a, miR-122, miR-126 | 10 mg/kg | ↑ | 3–3.5 | tumor from HCT-116 mouse xenograft | [22] |
| miR-143 | 10 mg/kg | ↑ | 3–3.5 | tumor from RKO mouse xenograft | [22] |
| miR-125a, miR-181a, miR-193b | 10 mg/kg | ↑ | 3.5–4 | tumor from HCT-116 mouse xenograft | [22] |
| let-7b | 10 mg/kg | ↑ | 4.5–5 | tumor from RKO mouse xenograft | [22] |
| miR-205 | 10 mg/kg | ↑ | 4.5–5 | tumor from HCT-116 mouse xenograft | [22] |
This entry is adapted from the peer-reviewed paper 10.3390/cancers14133056
References
- Yoontae Lee; Minju Kim; Jinju Han; Kyu-Hyun Yeom; Sanghyuk Lee; Sung Hee Baek; V Narry Kim; MicroRNA genes are transcribed by RNA polymerase II. The EMBO Journal 2004, 23, 4051-4060, 10.1038/sj.emboj.7600385.
- Yong Peng; Carlo M Croce; The role of MicroRNAs in human cancer. Signal Transduction and Targeted Therapy 2016, 1, 15004, 10.1038/sigtrans.2015.4.
- Leigh-Ann Macfarlane; Paul R. Murphy; MicroRNA: Biogenesis, Function and Role in Cancer. Current Genomics 2010, 11, 537-561, 10.2174/138920210793175895.
- Hiro-Oki Iwakawa; Yukihide Tomari; Life of RISC: Formation, action, and degradation of RNA-induced silencing complex. Molecular Cell 2021, 82, 30-43, 10.1016/j.molcel.2021.11.026.
- Rui Zhang; Ying Jing; Haiyang Zhang; Yahan Niu; Chang Liu; Jin Wang; Ke Zen; Chen-Yu Zhang; Donghai Li; Comprehensive Evolutionary Analysis of the Major RNA-Induced Silencing Complex Members.. Scientific Reports 2018, 8, 14189, 10.1038/s41598-018-32635-4.
- Andrew Jakymiw; Kaleb M. Pauley; Songqing Li; Keigo Ikeda; Shangli Lian; Theophany Eystathioy; Minoru Satoh; Marvin J. Fritzler; Edward Chan; The role of GW/P-bodies in RNA processing and silencing. Journal of Cell Science 2007, 120, 1317-1323, 10.1242/jcs.03429.
- Jidong Liu; Fabiola V. Rivas; James Wohlschlegel; John R. Yates; Roy Parker; Gregory J. Hannon; A role for the P-body component GW182 in microRNA function. Nature Cell Biology 2005, 7, 1261-1266, 10.1038/ncb1333.
- Muller Fabbri; Alessio Paone; Federica Calore; Roberta Galli; Eugenio Gaudio; Ramasamy Santhanam; Francesca Lovat; Paolo Fadda; Charlene Mao; Gerard J. Nuovo; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proceedings of the National Academy of Sciences 2012, 109, E2110-E2116, 10.1073/pnas.1209414109.
- George Adrian Calin; Calin Dan Dumitru; Masayoshi Shimizu; Roberta Bichi; Simona Zupo; Evan Noch; Hansjuerg Aldler; Sashi Rattan; Michael Keating; Kanti Rai; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences 2002, 99, 15524-15529, 10.1073/pnas.242606799.
- G A Calin; C M Croce; MicroRNAs and chromosomal abnormalities in cancer cells. Oncogene 2006, 25, 6202-6210, 10.1038/sj.onc.1209910.
- Yoji Hayashita; Hirotaka Osada; Yoshio Tatematsu; Hideki Yamada; Kiyoshi Yanagisawa; Shuta Tomida; Yasushi Yatabe; Katsunobu Kawahara; Yoshitaka Sekido; Takashi Takahashi; et al. A Polycistronic MicroRNA Cluster, miR-17-92, Is Overexpressed in Human Lung Cancers and Enhances Cell Proliferation. Cancer Research 2005, 65, 9628-9632, 10.1158/0008-5472.can-05-2352.
- Konstantinos J. Mavrakis; Andrew L. Wolfe; Elisa Oricchio; Teresa Palomero; Kim De Keersmaecker; Katherine McJunkin; Johannes Zuber; Taneisha James; Aly A. Khan; Christina S. Leslie; et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nature Cell Biology 2010, 12, 372-379, 10.1038/ncb2037.
- Lin Zhang; Jia Huang; Nuo Yang; Joel Greshock; Molly S. Megraw; Antonis Giannakakis; Shun Liang; Tara L. Naylor; Andrea Barchetti; Michelle R. Ward; et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proceedings of the National Academy of Sciences 2006, 103, 9136-9141, 10.1073/pnas.0508889103.
- George Adrian Calin; Cinzia Sevignani; Calin Dan Dumitru; Terry Hyslop; Evan Noch; Sai Yendamuri; Masayoshi Shimizu; Sashi Rattan; Florencia Bullrich; Massimo Negrini; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proceedings of the National Academy of Sciences 2004, 101, 2999-3004, 10.1073/pnas.0307323101.
- Kathryn A. O'Donnell; Erik A. Wentzel; Karen I. Zeller; Chi Dang; Joshua T. Mendell; c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839-843, 10.1038/nature03677.
- Tsung-Cheng Chang; Duonan Yu; Yun-Sil Lee; Erik A Wentzel; Dan E Arking; Kristin M West; Chi V Dang; Andrei Thomas-Tikhonenko; Joshua T Mendell; Widespread microRNA repression by Myc contributes to tumorigenesis. Nature Genetics 2007, 40, 43-50, 10.1038/ng.2007.30.
- Paloma Del C. Monroig; George A. Calin; MicroRNA and Epigenetics: Diagnostic and Therapeutic Opportunities. Current Pathobiology Reports 2013, 1, 43-52, 10.1007/s40139-013-0008-9.
- J. Michael Thomson; Martin Newman; Joel S. Parker; Elizabeth M. Morin-Kensicki; Tricia Wright; Scott M. Hammond; Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes & Development 2006, 20, 2202-2207, 10.1101/gad.1444406.
- Yoko Karube; Hisaaki Tanaka; Hirotaka Osada; Shuta Tomida; Yoshio Tatematsu; Kiyoshi Yanagisawa; Yasushi Yatabe; Junichi Takamizawa; Shinichiro Miyoshi; Tetsuya Mitsudomi; et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Science 2005, 96, 111-115, 10.1111/j.1349-7006.2005.00015.x.
- Jeffrey S. Dome; Max J. Coppes; Recent advances in Wilms tumor genetics. Current Opinion in Pediatrics 2002, 14, 5-11, 10.1097/00008480-200202000-00002.
- Ge Shan; Yujing Li; Junliang Zhang; Wendi Li; Keith E Szulwach; Ranhui Duan; Mohammad A Faghihi; Ahmad M Khalil; LiangHua Lu; Zain Paroo; et al. A small molecule enhances RNA interference and promotes microRNA processing. Nature Biotechnology 2008, 26, 933-940, 10.1038/nbt.1481.
- Sonia Melo; Alberto Villanueva; Catia Moutinho; Veronica Davalos; Riccardo Spizzo; Cristina Ivan; Simona Rossi; Fernando Setien; Oriol Casanovas; Laia Simo-Riudalbas; et al. Small molecule enoxacin is a cancer-specific growth inhibitor that acts by enhancing TAR RNA-binding protein 2-mediated microRNA processing. Proceedings of the National Academy of Sciences 2011, 108, 4394-4399, 10.1073/pnas.1014720108.
- Claudio De Vito; Nicolo Riggi; Sandrine Cornaz; Mario-Luca Suvà; Karine Baumer; Paolo Provero; Ivan Stamenkovic; A TARBP2-Dependent miRNA Expression Profile Underlies Cancer Stem Cell Properties and Provides Candidate Therapeutic Reagents in Ewing Sarcoma. Cancer Cell 2012, 21, 807-821, 10.1016/j.ccr.2012.04.023.
- Elsa J. Sousa; Inês Graça; Tiago Baptista; Filipa Q. Vieira; Carlos Palmeira; Rui Henrique; Carmen Jerónimo; Enoxacin inhibits growth of prostate cancer cells and effectively restores microRNA processing. Epigenetics 2013, 8, 548-558, 10.4161/epi.24519.
- Julia Ramírez-Moya; León Wert-Lamas; Garcilaso Riesco-Eizaguirre; Pilar Santisteban; Impaired microRNA processing by DICER1 downregulation endows thyroid cancer with increased aggressiveness. Oncogene 2019, 38, 5486-5499, 10.1038/s41388-019-0804-8.
- Ubaldo Gioia; Sofia Francia; Matteo Cabrini; Silvia Brambillasca; Flavia Michelini; Corey W. Jones-Weinert; Fabrizio D’Adda Di Fagagna; Pharmacological boost of DNA damage response and repair by enhanced biogenesis of DNA damage response RNAs. Scientific Reports 2019, 9, 6460, 10.1038/s41598-019-42892-6.
- Nathan S. Abell; Marvin Mercado; Tatiana Cañeque; Raphaël Rodriguez; Blerta Xhemalce; Click Quantitative Mass Spectrometry Identifies PIWIL3 as a Mechanistic Target of RNA Interference Activator Enoxacin in Cancer Cells. Journal of the American Chemical Society 2017, 139, 1400-1403, 10.1021/jacs.6b11751.
- Haruna Yamashiro; Mikiko C. Siomi; PIWI-Interacting RNA in Drosophila: Biogenesis, Transposon Regulation, and Beyond. Chemical Reviews 2017, 118, 4404-4421, 10.1021/acs.chemrev.7b00393.
- Lei Jiang; Wen-Jun Wang; Zhan-Wu Li; Xiao-Zhou Wang; Downregulation of Piwil3 suppresses cell proliferation, migration and invasion in gastric cancer. Cancer Biomarkers 2017, 20, 499-509, 10.3233/CBM-170324.
- Lan Li; Chaohui Yu; Hengjun Gao; Youming Li; Argonaute proteins: potential biomarkers for human colon cancer. BMC Cancer 2010, 10, 38-38, 10.1186/1471-2407-10-38.
- Shiguang Cao; Ruiying Sun; Wei Wang; Xia Meng; Yuping Zhang; Na Zhang; Shuanying Yang; RNA helicase DHX9 may be a therapeutic target in lung cancer and inhibited by enoxacin.. American journal of translational research 2017, 9, 674-682, .
- Chih-Hung Chou; Nai-Wen Chang; Sirjana Shrestha; Sheng-Da Hsu; Yu-Ling Lin; Wei-Hsiang Lee; Chi-Dung Yang; Hsiao-Chin Hong; Ting-Yen Wei; Siang-Jyun Tu; et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Research 2015, 44, D239-D247, 10.1093/nar/gkv1258.
- Yonit Hoffman; Yitzhak Pilpel; Moshe Oren; microRNAs and Alu elements in the p53-Mdm2-Mdm4 regulatory network. Journal of Molecular Cell Biology 2014, 6, 192-197, 10.1093/jmcb/mju020.
- Georgios Valianatos; Barbora Valcikova; Kateřina Growková; Amandine Verlande; Jitka Mlcochova; Lenka Radova; Monika Stetkova; Michaela Vyhnakova; Ondrej Slaby; Stjepan Uldrijan; et al. A small molecule drug promoting miRNA processing induces alternative splicing of MdmX transcript and rescues p53 activity in human cancer cells overexpressing MdmX protein. PLOS ONE 2017, 12, e0185801, 10.1371/journal.pone.0185801.
- Taylor C. Vracar; Jian Zuo; Jeongsu Park; Demyana Azer; Christy Mikhael; Sophia A. Holliday; Dontreyl Holsey; Guanghong Han; Lindsay VonMoss; John K. Neubert; et al. Enoxacin and bis-enoxacin stimulate 4T1 murine breast cancer cells to release extracellular vesicles that inhibit osteoclastogenesis. Scientific Reports 2018, 8, 16182, 10.1038/s41598-018-34698-9.
- Alicja Chrzanowska; Marta Struga; Piotr Roszkowski; Michał Koliński; Sebastian Kmiecik; Karolina Jałbrzykowska; Anna Zabost; Joanna Stefańska; Ewa Augustynowicz-Kopeć; Małgorzata Wrzosek; et al. The Effect of Conjugation of Ciprofloxacin and Moxifloxacin with Fatty Acids on Their Antibacterial and Anticancer Activity. International Journal of Molecular Sciences 2022, 23, 6261, 10.3390/ijms23116261.
- Alicja Chrzanowska; Piotr Roszkowski; Anna Bielenica; Wioletta Olejarz; Karolina Stępień; Marta Struga; Anticancer and antimicrobial effects of novel ciprofloxacin fatty acids conjugates. European Journal of Medicinal Chemistry 2019, 185, 111810, 10.1016/j.ejmech.2019.111810.
- H F Dvorak; Leaky tumor vessels: consequences for tumor stroma generation and for solid tumor therapy.. Progress in clinical and biological research 1990, 354A, 317-30, .
- Jr-Shiuan Yang; Eric C. Lai; Alternative miRNA Biogenesis Pathways and the Interpretation of Core miRNA Pathway Mutants. Molecular Cell 2011, 43, 892-903, 10.1016/j.molcel.2011.07.024.
- Thomas Maurin; Demián Cazalla; Jr-Shiuan Yang; Diane Bortolamiol-Becet; Eric C. Lai; RNase III-independent microRNA biogenesis in mammalian cells. RNA 2012, 18, 2166-2173, 10.1261/rna.036194.112.
- Jr-Shiuan Yang; Eric C. Lai; Dicer-independent, Ago2-mediated microRNA biogenesis in vertebrates. Cell Cycle 2010, 9, 4455-4460, 10.4161/cc.9.22.13958.
- Yong Xu; Fang Fang; Jiayou Zhang; Sajni Josson; William H. St. Clair; Daret K. St. Clair; miR-17* Suppresses Tumorigenicity of Prostate Cancer by Inhibiting Mitochondrial Antioxidant Enzymes. PLOS ONE 2010, 5, e14356, 10.1371/journal.pone.0014356.
- Munekazu Yamakuchi; Marcella Ferlito; Charles J. Lowenstein; miR-34a repression of SIRT1 regulates apoptosis. Proceedings of the National Academy of Sciences 2008, 105, 13421-13426, 10.1073/pnas.0801613105.
- Wei Wei; Yang Yang; Jian Cai; Kai Cui; Rong Xian Li; Huan Wang; Xiujuan Shang; Dong Wei; MiR-30a-5p Suppresses Tumor Metastasis of Human Colorectal Cancer by Targeting ITGB3. Cellular Physiology and Biochemistry 2016, 39, 1165-1176, 10.1159/000447823.
- Jian Zhang; Yongkang Zhang; Xiaoyun Li; Hongbo Wang; Quan Li; Xiaofeng Liao; MicroRNA-212 inhibits colorectal cancer cell viability and invasion by directly targeting PIK3R3. Molecular Medicine Reports 2017, 16, 7864-7872, 10.3892/mmr.2017.7552.
- Roberto Gambari; Eleonora Brognara; Demetrios A. Spandidos; Enrica Fabbri; Targeting oncomiRNAs and mimicking tumor suppressor miRNAs: New trends in the development of miRNA therapeutic strategies in oncology (Review). International Journal of Oncology 2016, 49, 5-32, 10.3892/ijo.2016.3503.
- Maryam Nurzadeh; Mahsa Naemi; Shahrzad Sheikh Hasani; A comprehensive review on oncogenic miRNAs in breast cancer. Journal of Genetics 2021, 100, 1-21, 10.1007/s12041-021-01265-7.
- Elisa Penna; Francesca Orso; Daniela Taverna; miR-214 as a Key Hub that Controls Cancer Networks: Small Player, Multiple Functions. Journal of Investigative Dermatology 2015, 135, 960-969, 10.1038/jid.2014.479.
- Mengru Cao; Masahiro Seike; Chie Soeno; Hideaki Mizutani; Kazuhiro Kitamura; Yuji Minegishi; Rintaro Noro; Akinobu Yoshimura; Li Cai; Akihiko Gemma; et al. MiR-23a regulates TGF-β-induced epithelial-mesenchymal transition by targeting E-cadherin in lung cancer cells. International Journal of Oncology 2012, 41, 869-875, 10.3892/ijo.2012.1535.
- Monireh Khordadmehr; Roya Shahbazi; Sanam Sadreddini; Behzad Baradaran; miR‐193: A new weapon against cancer. Journal of Cellular Physiology 2019, 234, 16861-16872, 10.1002/jcp.28368.
- Arata Itoh; David Adams; Wenting Huang; Yuehong Wu; Kritika Kachapati; Kyle J. Bednar; Patrick S. C. Leung; Weici Zhang; Richard A. Flavell; M. Eric Gershwin; et al. Enoxacin Up‐Regulates MicroRNA Biogenesis and Down‐Regulates Cytotoxic CD8 T‐Cell Function in Autoimmune Cholangitis. Hepatology 2021, 74, 835-846, 10.1002/hep.31724.
- Neil R. Smalheiser; Hui Zhang; Yogesh Dwivedi; Enoxacin Elevates MicroRNA Levels in Rat Frontal Cortex and Prevents Learned Helplessness. Frontiers in Psychiatry 2014, 5, 6, 10.3389/fpsyt.2014.00006.
- Xinbo Huang; Zhicong Chen; Yuchen Liu; RNAi-mediated control of CRISPR functions. Theranostics 2020, 10, 6661-6673, 10.7150/thno.44880.
- miRNA’s Nomenclature . miRBase. Retrieved 2022-7-1