Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
African Americans (AA) are disproportionately burdened by metabolic diseases. While largely unexplored between Caucasian (C) and AA, differences in mitochondrial bioenergetics may provide crucial insight to mechanisms for increased susceptibility to metabolic diseases. AA display lower total energy expenditure and resting metabolic rate compared to C, but paradoxically have a higher amount of skeletal muscle mass, suggestive of inherent energetic efficiency differences between these races.
- insulin
- African American
- mitochondria
1. Introduction
African Americans have disproportionately higher rates of obesity that persist across all ages and genders [1]. According to the Office of Minority Health, non-Hispanic African American (AA) adults are 1.3 times (47.9% vs. 37.4%) more likely to have obesity compared to non-Hispanic Caucasians (C) [1]. Likewise, AA children and adolescents are 1.4 times more likely to have obesity compared to C children and adolescents [1]. Furthermore, the obesity gap amplifies across gender with AA women being 1.5 times more likely to have obesity compared to C women. In fact, 55% of all AA women in the US can be classified in the obese body mass index (BMI > 30) category [2]. A contributing factor to these racial disparities is likely multifactorial, but several hypotheses have been proposed. One such hypothesis is that overall lower physical activity underlies these disparities, as 50.3% of AA do not meet the federal physical activity guidelines compared to 38.9% of their C counterparts [1]. Though the influence of socioeconomic status is not to be understated, it is worth noting that this pattern persists across the middle- and lower-income levels, and in males, this discrepancy grows even further at high-income levels [3]. Moreover, both AA and C races follow a similar pattern where the level of education is inversely related to the prevalence of obesity; however, rates of obesity are persistently higher among AA women at any education level [3].
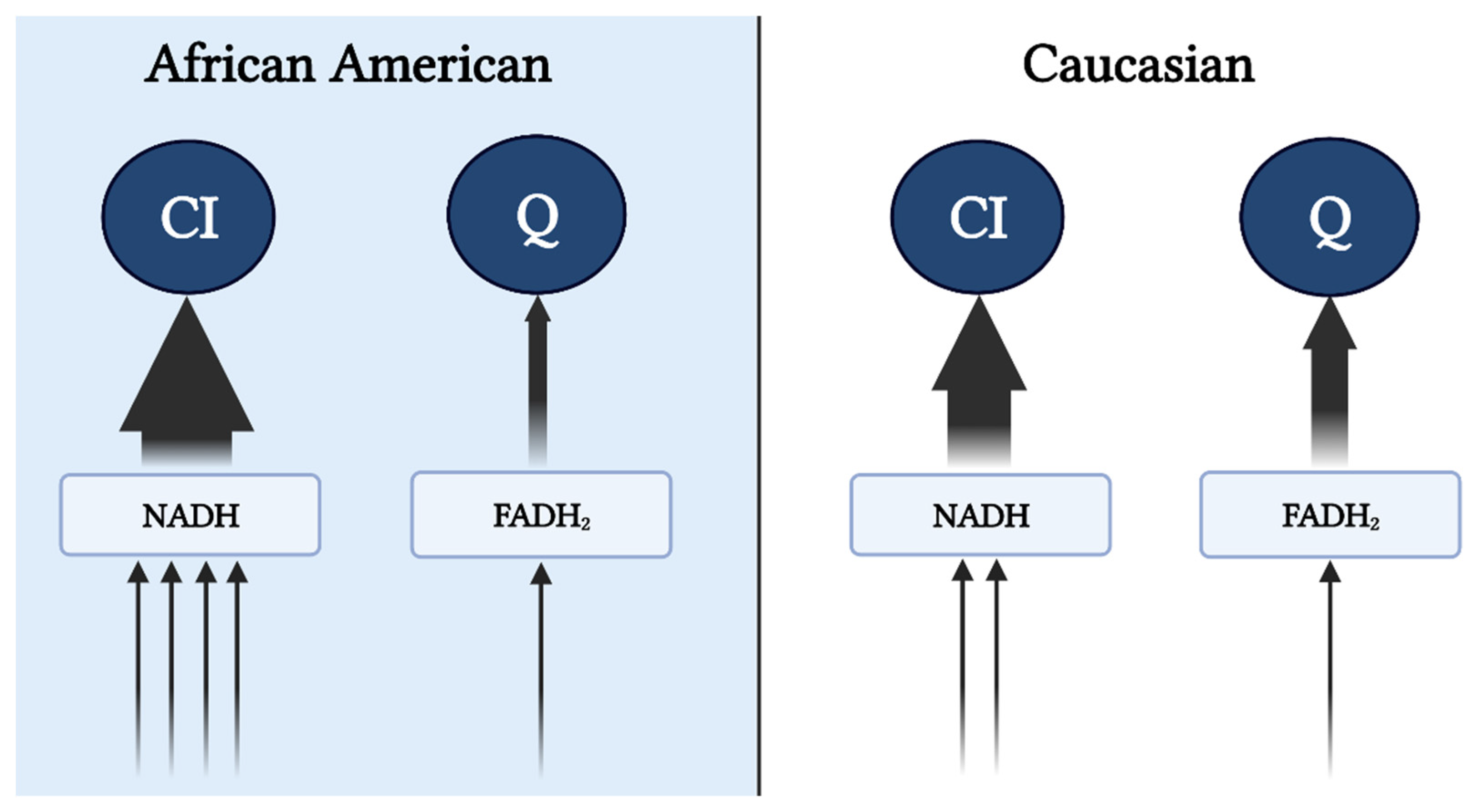 Figure 1. Preferential input of electrons into CI via NAD-linked substrates in AA. Heightened glycolytic flux ensues greater provision of NAD-linked pathways. From cytosol, NADH is brought into the mitochondrial matrix via malate-aspartate, G-3-P, and/or lactate shuttle(s) subsequently supporting the CI-linked respiration (described in the subsequent text). Supporting respiration through pyruvate, malate, and glutamate provides a 4:1 ratio (represented by the arrows) of NAD to FADH reduction with a complete TCA cycle. In contrast, Caucasians have a preferential respiration through succinate and fatty oxidation pathways resulting in a 2:1 reduction in NAD and FADH. This image was created using BioRender.com, accessed on 11 May 2022.
Figure 1. Preferential input of electrons into CI via NAD-linked substrates in AA. Heightened glycolytic flux ensues greater provision of NAD-linked pathways. From cytosol, NADH is brought into the mitochondrial matrix via malate-aspartate, G-3-P, and/or lactate shuttle(s) subsequently supporting the CI-linked respiration (described in the subsequent text). Supporting respiration through pyruvate, malate, and glutamate provides a 4:1 ratio (represented by the arrows) of NAD to FADH reduction with a complete TCA cycle. In contrast, Caucasians have a preferential respiration through succinate and fatty oxidation pathways resulting in a 2:1 reduction in NAD and FADH. This image was created using BioRender.com, accessed on 11 May 2022.
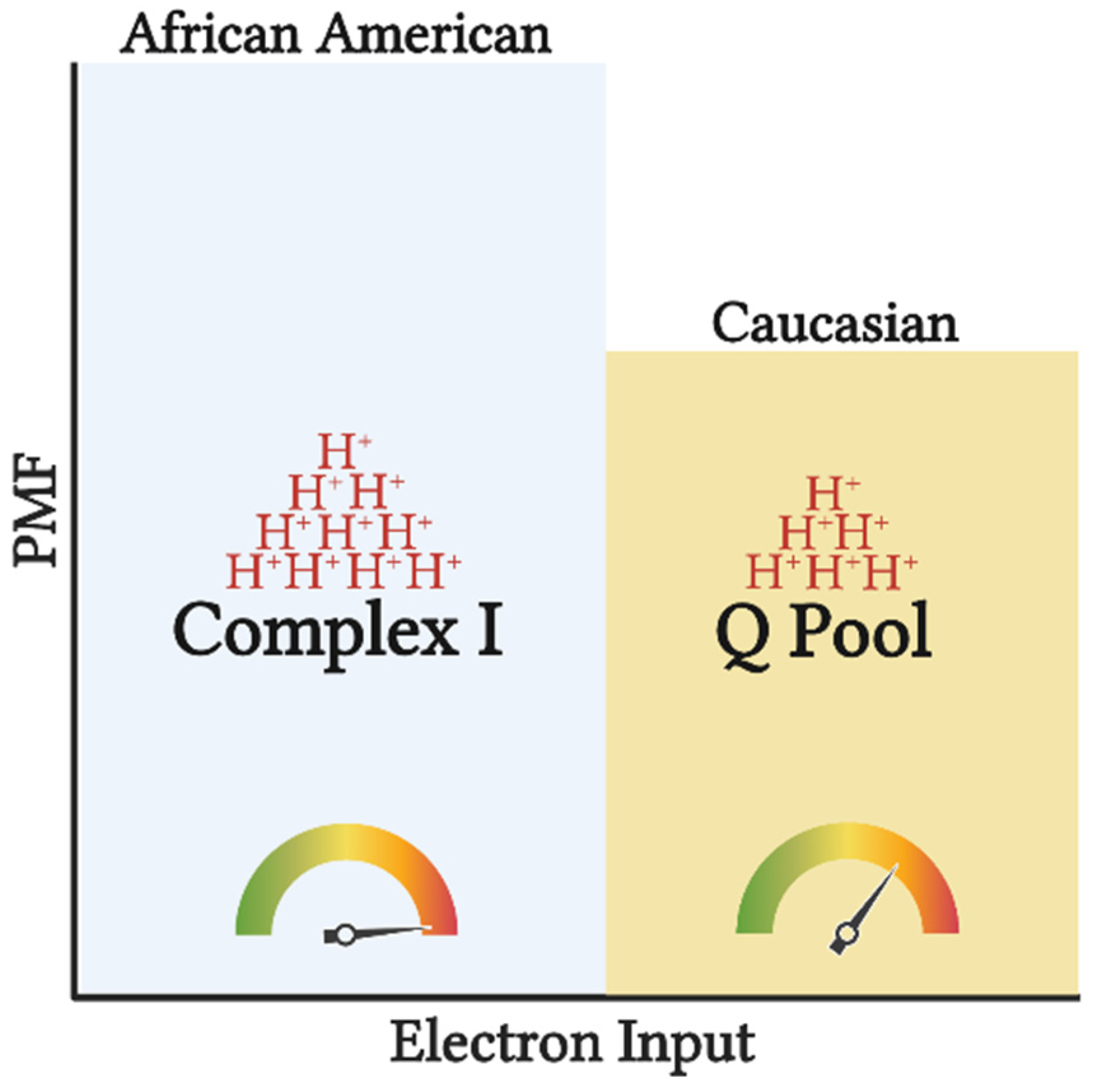 Figure 2. Tightly coupled mitochondrial respiration and NAD-linked substrate input potentiates greater efficiency (depicted by the gauge) in AA. Input of electrons into CI compared to the Q pool, will generate a greater H+ translocation across inner mitochondrial membrane (depicted by the H+ stacks) increasing the proton motive force (PMF), and subsequently ATP production. Such setup allows for greater ATP synthesis per substrate utilized, increasing the chances of overnutrition and subsequently obesity. This image was created using BioRender.com, accessed on 11 May 2022.
Figure 2. Tightly coupled mitochondrial respiration and NAD-linked substrate input potentiates greater efficiency (depicted by the gauge) in AA. Input of electrons into CI compared to the Q pool, will generate a greater H+ translocation across inner mitochondrial membrane (depicted by the H+ stacks) increasing the proton motive force (PMF), and subsequently ATP production. Such setup allows for greater ATP synthesis per substrate utilized, increasing the chances of overnutrition and subsequently obesity. This image was created using BioRender.com, accessed on 11 May 2022.
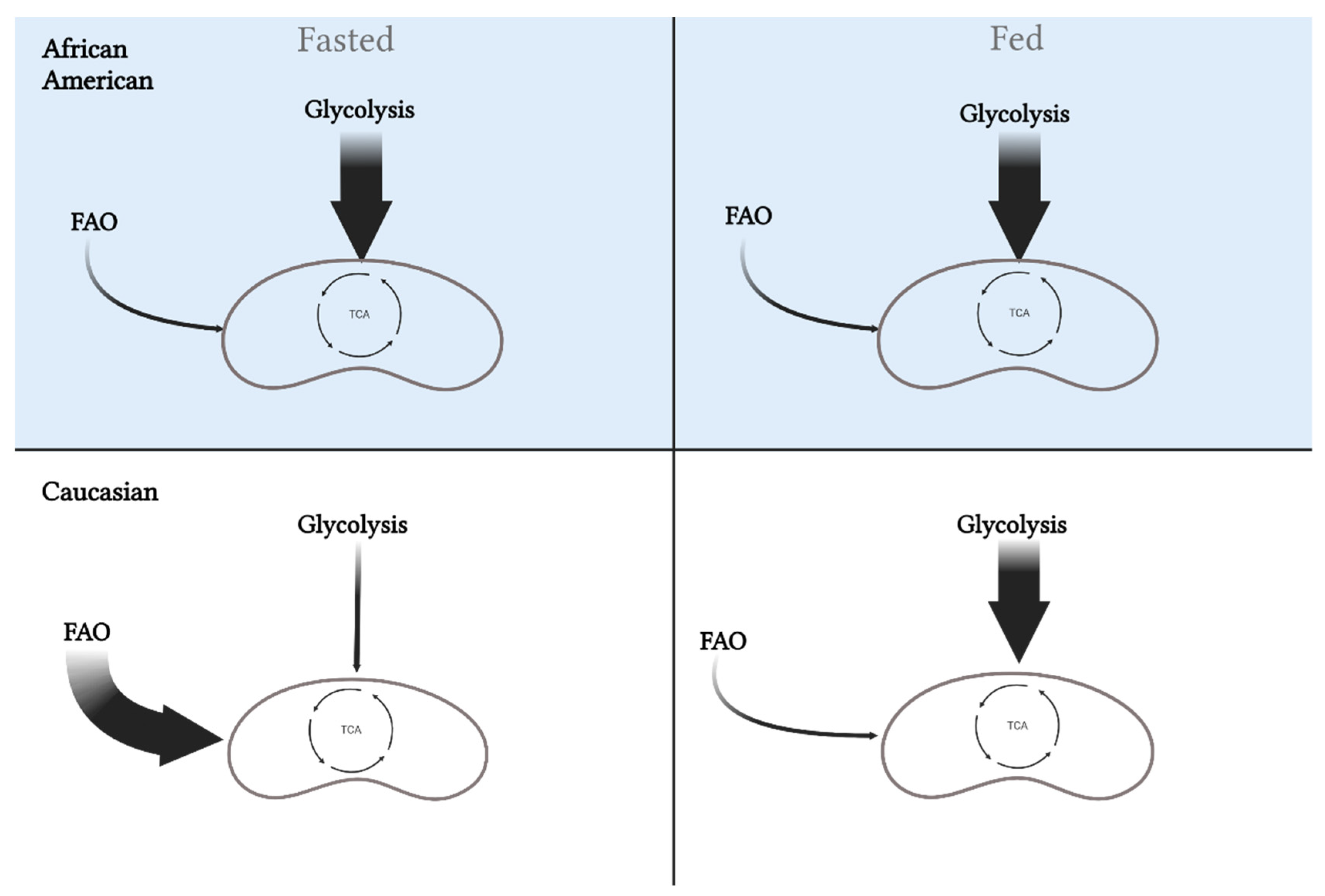 Figure 3. Differences in metabolic flexibility between African Americans and Caucasians. Metabolic flexibility is defined as the ability to switch between substrates (carbohydrates versus fat) in the fasted and fed state. African Americans rely primarily on glycolysis in the fasted state, which is maintained in the fed state. Alternatively, Caucasians will oxidize more fat in the fasted state and increase oxidization of carbohydrates in the fed state. These differences may play a role in the disparity for metabolic diseases between the races. This image was created using BioRender.com, accessed on 11 May 2022.
Figure 3. Differences in metabolic flexibility between African Americans and Caucasians. Metabolic flexibility is defined as the ability to switch between substrates (carbohydrates versus fat) in the fasted and fed state. African Americans rely primarily on glycolysis in the fasted state, which is maintained in the fed state. Alternatively, Caucasians will oxidize more fat in the fasted state and increase oxidization of carbohydrates in the fed state. These differences may play a role in the disparity for metabolic diseases between the races. This image was created using BioRender.com, accessed on 11 May 2022.
2. Mitochondrial Efficiency Increases Predisposition of African Americans to Obesity
2.1. Energy Expenditure
The primary cause of obesity is often attributed to an energy imbalance from energy consumed and expended. Thus, it is essential to consider energy expenditure and its impact in the development of obesity as a causative factor in the racial disparity in the development of obesity. For example, it has been well documented that both total energy expenditure (TEE) and resting metabolic rate (RMR) are lower in AA compared to C counterparts across multiple age groups and regardless of gender or obesity status [4][5]. Partially responsible for the lower RMR is a lower mass of organs with high metabolic rates (i.e., liver, heart, etc.) in AA compared to C [6]. However, as these organs themselves do not comprise most of the active metabolic tissue in the body, it is essential to consider the role of skeletal muscle in TEE. Skeletal muscle itself comprises about 40% of total body weight and thus contributes substantially to energy expenditure as it is a prominent constituent of RMR [7]. Furthermore, considering that RMR comprises ~65% of TEE, skeletal muscle mass plays an essential role in determining TEE [7][8]. Paradoxically, while AA seem to have a greater percentage of relative skeletal muscle mass, they exhibit lower RMR making them more susceptible to overnutrition [5][9]. As skeletal muscle metabolism is largely dictated by mitochondrial bioenergetics, it is important to note the mitochondrial DNA variations that exist between AA and C populations and their effect on RMR, TEE, and overall risk for metabolic disease [10][11]. When compared to common European haplogroups (i.e., H, JT), common African haplogroups (i.e., L0, L2) exhibit significantly lower RMR and TEE even after adjusting for lean mass [12]. Furthermore, it is noteworthy to recognize that even within common racial haplogroups (i.e., AA), RMR and TEE can vary significantly [12]. Accordingly, future studies should attempt to control for these variances whenever possible, as these factors can be influential of the findings and introduce variability within a group. Taken together, these data provide evidence that certain genetic components may help drive racial dichotomies in energy metabolism, ultimately, making AA more susceptible to overnutrition.
2.2. Mitochondrial “Substrate Preference”
Reduced mitochondrial content and respiratory capacity have been reported in AA women [13][14], which is accompanied by lower maximal coupled and uncoupled respiration rates that persist despite adjustments for oxidative fiber-type content [13]. Additionally, lower state 3 (ADP stimulated), state U (uncoupled), and state 4 (leak) respiration have been observed in permeabilized fibers after normalization to mitochondrial content [14]. While it is tempting to ascribe lower maximal coupled and uncoupled respiration to mitochondrial dysfunction, it must be noted that oxidative phosphorylation (OXPHOS) and electron transfer capacity (independent of ATP synthase) under non-physiological conditions (lack of control over adenylate pool) provide limited information regarding mitochondrial bioenergetic function. Furthermore, the assays used in these studies were performed using mixed substrates during high-resolution respirometry, which could misrepresent these findings as mixed substrates prohibit the delineation of “substrate preference”. For example, the lower capacity (JO2) observed in AA could be largely due to the preferential utilization of NAD-linked, rather than FAD-linked substrates. The increase in glycolytic flux and decrease in fatty acid oxidation in AA compared to C (see subsequent sections) would increase the availability of NADH and NAD-linked substrates (i.e., pyruvate), potentiating preferential input of electrons to complex I (Figure 1), rather than Q pool (via CII or electron-transferring flavoproteins). In this case, more protons (~10 if entering at CI versus ~6 if entering at CII) are pumped out per reduced oxygen, resulting in a lower oxygen turnover (JO2) but greater efficiency (ATP/O ratio), leading to greater ATP production per amount of substrate utilized. This is in line with the evidence that AA have shorter recovery times during isometric exercise, consistent with a higher ATP synthesis rate, suggesting a greater energetic efficiency in AA [15]. This notion in concert with the lower recorded energy expenditure suggests a greater metabolic efficiency in AA compared to C.
Figure 1. Preferential input of electrons into CI via NAD-linked substrates in AA. Heightened glycolytic flux ensues greater provision of NAD-linked pathways. From cytosol, NADH is brought into the mitochondrial matrix via malate-aspartate, G-3-P, and/or lactate shuttle(s) subsequently supporting the CI-linked respiration (described in the subsequent text). Supporting respiration through pyruvate, malate, and glutamate provides a 4:1 ratio (represented by the arrows) of NAD to FADH reduction with a complete TCA cycle. In contrast, Caucasians have a preferential respiration through succinate and fatty oxidation pathways resulting in a 2:1 reduction in NAD and FADH. This image was created using BioRender.com, accessed on 11 May 2022.It is feasible that evolutionary adaptations have occurred as a result of tropical climates and lower accessibility to nutrients to maximize the efficiency of nutrient use and yield the highest ATP free energy [16]. Furthermore, along with the thrifty phenotype hypothesis, the AA mitochondrial phenotype would support maximal ATP and minimal heat production per calorie utilized [17]. This tight coupling of mitochondrial respiration to drive ATP synthesis would ensure greater efficiency in the utilization of carbon backbones provided by oxidized substrates. Additionally, high ETS efficiency and/or preferential use of NAD-linked substrates yields a more polarized mitochondrial membrane potential that is in inverse relation to the electron flow (Figure 2). Such a phenotype would explain the lower oxygen consumption rate and manifested as a lower RMR. This adaptation would likely be advantageous in scenarios of low and inconsistent nutrient availability; however, tightly coupled mitochondria in the presence of readily available nutrients would predispose AA individuals to overnutrition and promote disproportionate rates of obesity between races in an energy affluent environment. It must be noted that lower mitochondrial content and oxidative capacity are only in part influential on the amount of substrate being oxidized. Considering that the energetic state is derived from the cellular redox state, ATP production will take precedence over oxidative capacity in directing cellular bioenergetics or energy transfer from substrate to ATP production. Simply stated, the amount of oxidized substrate is dictated by the energy demand, rather than the energy input into the system. This suggests that regardless of the change in oxidative capacity contingent upon the shift in mitochondrial content, mitochondrial efficiency is a major determinant of substrate oxidation. While the chances of overnutrition increase considering the lower energy expenditure in AA, such an effect would not explain the racial difference in IR in healthy subjects [18]. Interestingly, mitochondrial capacity was inversely related to peripheral insulin sensitivity in work carried out by DeLany and colleagues, suggesting that this mitochondrial phenotype is a contributing factor to IR in AA [13]. Similarly, lower mitochondrial capacity has been reported in type 2 diabetics (T2D), inversely correlated with glycemic control, and directly corelated with metabolic inflexibility [19][20]. Accordingly, while in AA lower oxidative capacity might not stem from mitochondrial functional impairments, it can still have a role in increasing their predisposition to IR.
Figure 2. Tightly coupled mitochondrial respiration and NAD-linked substrate input potentiates greater efficiency (depicted by the gauge) in AA. Input of electrons into CI compared to the Q pool, will generate a greater H+ translocation across inner mitochondrial membrane (depicted by the H+ stacks) increasing the proton motive force (PMF), and subsequently ATP production. Such setup allows for greater ATP synthesis per substrate utilized, increasing the chances of overnutrition and subsequently obesity. This image was created using BioRender.com, accessed on 11 May 2022.3. Mitochondrial Phenotype Increases Predisposition of African Americans to Insulin Resistance
3.1. Insulin Resistance and Metabolic (In)Flexibility
Skeletal muscle is responsible for ~80% of postprandial glucose uptake and yet, AA paradoxically display higher rates of IR despite having higher skeletal muscle mass [11]. Predictive measures of IR, such as disposition and β-cell function indices, are noted to be higher in AA [11]. Furthermore, in conditions of normal glucose tolerance, evidence of upregulated β-cell function as well as a rightward shift in the glucose allostasis model (relationship between insulin sensitivity and acute insulin response) is present [11]. Thus, the chronic hyperinsulinemia observed in AA must be considered as another factor in this racial disparity due to its strong association with obesity and other metabolic diseases (i.e., T2D) [12].
All factors considered, it seems that impaired glucose disposal at peripheral tissues lie at the core of racial disparities in glycemic control [11]. This paradigm observed in normoglycemic AA parallels a similar model of metabolic abnormalities that occur with IR in C [13]. Regarding IR, the progressive increase in insulin secretion in response to decreased peripheral tissue sensitivity leads to β-cell “stress” and subsequently exhaustion, demise, and dedifferentiation [14]. While IR is traditionally negative in terms of cardiometabolic disease with C, in the AA population, and in the context of inherent increased insulin output, it may serve as a protective mechanism against weight gain and obesity-related diseases (i.e., less fuel availability intracellularly for storage) [12]. However, genetically driven impairments in glucose uptake could predispose AA to a greater risk of T2D development as well and be further amplified in situations of overnutrition [12].
Metabolic flexibility has been implicated as a major predictor of insulin resistance and metabolic disease. Under conditions of energy metabolism unaffected by metabolic diseases, a metabolically flexible state can be characterized by mitochondria alternating between substrates based on physiologic needs and nutritional states. In contrast, in the state of metabolic inflexibility, crosstalk and regulation of substrate choice and substrate oxidation via metabolic and cell signaling events are dampened. Metabolic inflexibility, as observed in subjects with obesity and T2D, displays itself as a lack of substrate switching across changes in physiological states—a switch from the fasted state to a fed state and back [21].
Metabolic inflexibility and consequently the lack of substrate switching itself could have implications in the racial dichotomy between AA and C. Lower fat but higher carbohydrate oxidation and overall metabolic inflexibility have been reported in AA in the postabsorptive state and during high-fat and low-fat diets, euglycemic clamps, and epinephrine-induced lipolysis [22]. An inability to switch between substrates during eucaloric macronutrient-manipulated diets suggests that lean AA participants have an inherent preference for carbohydrate use. Moreover, it is worth noting that AA women display greater fasting plasma insulin during the low-fat and high-fat diet, which was contrary to C participants [22]. Additionally, contrary to C participants, with insulin administration during the euglycemic clamp, AA failed to suppress fatty acid oxidation. Similar outcomes were observed during the high-fat diet where there was no change in fat oxidation nor carbohydrate oxidation, suggesting that the fatty-acid-induced inhibition of glucose metabolism (i.e., Randle cycle) is not as prominent in AA [22]. Together, these findings in AA women with either normal or obese BMI suggest that there is preferential carbohydrate oxidation and that the manipulation of the physiologic environment via dietary habits is not sufficient to overcome these intrinsic biological metabolic differences.
Metabolic inflexibility is influenced by the potential for lipid and carbohydrate oxidation determined by different muscle fiber-type compositions, and accordingly, variance in myocellular metabolic processes. Compared to C, AA display a lower proportion of insulin-sensitive type I muscle fibers geared towards oxidative metabolism combined with the higher proportion of type II glycolytic fibers, which results in a lower capacity for lipid oxidation at rest and during low-intensity exercise [22][23]. Furthermore, in normal weight and AA with obesity, these specific fiber-type distributions are associated with lower training adaptations, reduced oxidative capacity, and glycemic control [24][25][26][27][28][29]. Such fiber-type differences have been previously correlated with decreased insulin sensitivity and could influence the difference in obesity and T2D rates between these two races [25][29][30][31][32]. Specifically, higher glycolytic to oxidative muscle fiber-type ratio will potentiate higher glycolytic flux and could inhibit lipid oxidation via elevated production of malonyl CoA (i.e., reverse Randle cycle) [33]. Together, this altered substrate flux stemming from muscle fiber-type and innate metabolic differences potentiate metabolic inflexibility in AA (Figure 3).
Figure 3. Differences in metabolic flexibility between African Americans and Caucasians. Metabolic flexibility is defined as the ability to switch between substrates (carbohydrates versus fat) in the fasted and fed state. African Americans rely primarily on glycolysis in the fasted state, which is maintained in the fed state. Alternatively, Caucasians will oxidize more fat in the fasted state and increase oxidization of carbohydrates in the fed state. These differences may play a role in the disparity for metabolic diseases between the races. This image was created using BioRender.com, accessed on 11 May 2022.3.2. Muscle Enzymatic Differences and Lower Fatty Acid Oxidation Potential
The previously described metabolic inflexibility of AA might be further influenced by phenotypic differences in metabolic pathways [24][27]. In muscle biopsies of the vastus lateralis, biochemical analyses showed lower oxidative and greater glycolytic pathway enzymatic activities in AA compared to C [27]. Sedentary AA males had a lower percentage of type I and a higher percentage of type IIa muscle fibers, which paralleled the higher phosphogenic and glycolytic enzyme activities when compared to C counterparts [27]. Additionally, as a surrogate measure of pathway predominance, Ama and colleagues reported a ~32% lower phosphofructokinase to oxoglutarate dehydrogenase ratio in C when compared to AA males. At the level of the TCA cycle, no differences were found in citrate synthase (CS), malate dehydrogenase, and oxoglutarate dehydrogenase between AA and C [27][33]. However, it is worth noting that CS was assessed in muscle from women with morbid obesity, which could be a reason for the lack of difference, considering that lower CS activity is associated with obesity [34]. Although CS is often a surrogate marker of mitochondrial content, observations of lower mitochondrial content in AA when compared to C would suggest an inherently lower CS content, but not CS activity per se [14][33].
While DeLany et al. observed lower succinate dehydrogenase in non-obese sedentary AA women when compared to their C counterparts [13], β-oxidation was not altered, as no differences in β-hydroxy acyl CoA were found [27][33]. Furthermore, lower mitochondrial and microsomal acyl-CoA synthetase activity was seen suggesting that decrements in oxidation stem from a lower fatty acid activation via acyl CoA synthetase [33]. This notion is supported by observed lower fatty acid oxidation in rectus abdominus strips and vastus lateralis muscle homogenates of AA compared to C [24][33]. In AA with normal and obese BMI classifications, there is lower oxidation of palmitate but not activated fatty acids (palmitoyl-CoA) [24]. Additionally, no differences in palmitate-carnitine were observed suggesting no difference at the level of carnitine-acylcarnitine translocase [24]. While Cortright et al. saw significant differences in subjects with obesity, the difference in palmitate oxidation between lean AA and C was only trending. This suggests that innate differences only predispose AA to decrements in fatty acid oxidation, and that significant effects might be dependent on and exacerbated with metabolic stress (i.e., overnutrition, obesity).
In accordance with the reverse Randle cycle theory, higher glucose uptake under conditions of innate hyperinsulinemia and higher glycolytic fiber-type content may be in part responsible for the downregulation of fatty acid oxidation [35]. Furthermore, lower succinate dehydrogenase would lead to a backup in the TCA cycle allowing for the accumulation and efflux of citrate into the cytosol. In skeletal muscle, this would lead to the accumulation of malonyl-CoA inhibiting CPT-1 and the rerouting of long-chain fatty acyl moieties towards esterification and storage [35]. The proposition of CPT-1 inhibition is unlikely based on the studies by Privette et al. [33] and Cortright et al. [24]; however, in these studies, muscle homogenate oxidation was measured in the presence of palmitate only, neglecting the potential effect of higher glucose flux. Additionally, the measures obtained do not mimic the postprandial state in which glucose availability and influx into the cell is highest and holds the greatest potential to influence substrate preference. The influence of glucose and dietary glycemic load on fat metabolism in AA has been highlighted in a recent review by Gower and Fowler [18]. The researchers suggest that individuals with high postprandial insulin have a chronic “brake” on lipid metabolism predisposing them to obesity. Accordingly, AA compared to C lost significantly more fat mass on a low glycemic compared to low-fat diet. Such evidence would suggest that the combination of genetic hyperinsulinemia and high glucose availability potentiate glycolytic flux and lipid backup considering that fat and carbohydrate oxidation are mutually inhibitory. Lastly, more efficient OXPHOS and lower FAO increases the susceptibility of fatty acid accumulation with overnutrition. Together, such evidence puts fatty acid accumulation in the “spotlight” when it comes to the impairments in insulin sensitivity; however, this might not be the case when it comes to AA.
3.3. Ectopic Lipid Accumulation Is Not a Predictor of Insulin Resistance in AA
Lower fatty acid oxidation in parallel with metabolic inflexibility facilitates the partitioning of lipids towards ectopic accumulation. Excessive total and ectopic fat deposition are known to contribute to the development of IR. Interestingly, while patterns of ectopic fat deposition differ between AA and C, their contribution to IR is not clearly demonstrated. AA have a trend towards higher intermuscular adipose tissue and this difference becomes significant with increased adiposity; however, it is not a strong predictor of insulin sensitivity in AA [36][37][38][39]. Additionally, a similar intramyocellular lipid content has been observed between AA and C in a recent meta-analysis across all BMIs, which suggests that the total accumulation of the intramyocellular lipids is not a major contributor to differences in peripheral IR between the races [40]. Nonetheless, while intramyocellular lipid content is correlated with insulin sensitivity in C, there is no association between these parameters in healthy and overweight AA subjects, suggesting that other fat depots or specific lipid species take precedent in influencing insulin sensitivity in AA [41][42]. Compared to C, it has been shown that AA have significantly lower visceral adiposity but significantly greater subcutaneous adipose depots [40][43]. Furthermore, both visceral and subcutaneous adipose tissue have been correlated with IR in both healthy and glucose-intolerant AA subjects [44]. These observations suggest that peripheral IR in AA might be subject to the composition of specific muscular lipid species rather than quantities considering that the associations between ectopic lipid accumulation and insulin sensitivity in either healthy or AA with obesity are weak.
Intramyocellular lipids are composed of predominantly triacylglycerols, but also include diacylglycerols, ceramides, and long-chain fatty acyl-CoAs. Of these lipid species, ceramides and diacylglycerols have been implicated as drivers of lipotoxicity-induced IR [41]. While there is a lack of research regarding intramyocellular lipid profiling, plasma levels of C16:0 ceramides are shown to be similar between healthy AA and C [45]. Moreover, AA had trends towards higher C18:0, C18:1, C22:0, and C24:0 levels [45]. This trend would imply higher C22:0/C16:0 and C24:0/C16:0 ratios which have been associated with an inverse relationship with all-cause mortality and coronary heart disease [45][46]. Similarly, AA subjects have a better ceramide ratio (C18:1/C18:0)/(C18:1/C16:0), which has been implicated as an independent marker predictive of T2D even after adjusting for BMI, fasting glucose, and HbA1c [45][47]. In addition, while a trend towards overall higher total ceramide content was observed in healthy AA, the opposite was found in AA with metabolic disease potentially due to the lack of adjustment for any of the reported confounding variables (i.e., medication). Finally, the only significant difference between healthy groups was higher levels of sphingosine 1-phosphate (S1P) in AA [45]. However, contradictory findings regarding the involvement of S1P in IR exist making it hard to conclude if there is an association between insulin sensitivity and these lipid species in AA [42].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10061456
References
- Office of Minority Health. Available online: https://www.cdc.gov/nchs/nhis/shs/tables.htm (accessed on 27 April 2022).
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of obesity among adults and youth: United States, 2015–2016. NCHS Data Brief 2015, 288, 1–8.
- Ogden, C.; Lamb, M.; Carroll, M.; Flegal, K. Obesity and socioeconomic status in adults: United States, 2005–2008. NCHS Data Brief 2010, 50, 1–8.
- Gannon, B.; Dipietro, L.; Poehlman, E.T. Do African Americans have lower energy expenditure than Caucasians? Int. J. Obes. Relat. Metab. Disord. 2000, 24, 4–13.
- Katzmarzyk, P.T.; Most, J.; Redman, L.M.; Rood, J.; Ravussin, E. Energy expenditure and substrate oxidation in White and African American young adults without obesity. Eur. J. Clin. Nutr. 2018, 72, 920–922.
- Gallagher, D.; Albu, J.; He, Q.; Heshka, S.; Boxt, L.; Krasnow, N.; Elia, M. Small organs with a high metabolic rate explain lower resting energy expenditure in African American than in white adults 2. Am. J. Clin. Nutr. 2006, 83, 1062–1067.
- Zurlo, F.; Larson, K.; Bogardus, C.; Ravussin, E. Skeletal muscle metabolism is a major determinant of resting energy expenditure. J. Clin. Investig. 1990, 86, 1423–1427.
- Browning, M.G.; Evans, R.K. The contribution of fat-free mass to resting energy expenditure: Implications for weight loss strategies in the treatment of adolescent obesity. Int. J. Adolesc. Med. Health 2015, 27, 241–246.
- Heymsfield, S.B.; Peterson, C.M.; Thomas, D.M.; Heo, M.; Schuna, J.M. Why are there race/ethnic differences in adult body mass index-adiposity relationships? A quantitative critical review. Obes. Rev. 2016, 17, 262–275.
- Dunham-Snary, K.J.; Ballinger, S.W. Mitochondrial genetics and obesity: Evolutionary adaptation and contemporary disease susceptibility. Free Radic. Biol. Med. 2013, 65, 1229–1237.
- Kenney, M.; Falatoonzadeh, P.; Atilano, S.; Chwa, M.; Cáceres-del-Carpio, J.; Malik, D.; Boyer, D.; Nesburn, A.; Kuppermann, B. African-origin Mitochondrial DNA Variants as a Contributing Factor to Susceptibilities for Diabetes and Age-related Diseases. Int. J. Diabetes Clin. Res. 2016, 3, 53.
- Tranah, G.J.; Manini, T.M.; Lohman, K.K.; Nalls, M.A.; Kritchevsky, S.; Newman, A.B.; Harris, T.B.; Miljkovic, I.; Biffi, A.; Cummings, S.R.; et al. Mitochondrial DNA variation in human metabolic rate and energy expenditure. Mitochondrion 2011, 11, 855–861.
- DeLany, J.P.; Dubé, J.J.; Standley, R.A.; Distefano, G.; Goodpaster, B.H.; Stefanovic-Racic, M.; Coen, P.M.; Toledo, F.G.S. Racial differences in peripheral insulin sensitivity and mitochondrial capacity in the absence of obesity. J. Clin. Endocrinol. Metab. 2014, 99, 4307–4314.
- Toledo, F.G.S.; Dubé, J.J.; Goodpaster, B.H.; Stefanovic-Racic, M.; Coen, P.M.; DeLany, J.P. Mitochondrial respiration is associated with lower energy expenditure and lower aerobic capacity in African American women. Obesity 2018, 26, 903–909.
- Sirikul, B.; Gower, B.A.; Hunter, G.R.; Larson-Meyer, D.E.; Newcomer, B.R.; Sirikul, B.A.; Gower, G.R.; Hunter, D.E.; Larson-Meyer, B. Relationship between insulin sensitivity and in vivo mitochondrial function in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2006, 291, 724–728.
- Coskun, P.E.; Ruiz-Pesini, E.; Wallace, D.C. Control region mtDNA variants: Longevity, climatic adaptation, and a forensic conundrum. Proc. Natl. Acad. Sci. USA 2003, 100, 2174–2176.
- Ruiz-Pesini, E.; Mishmar, D.; Brandon, M.; Procaccio, V.; Wallace, D.C. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 2004, 303, 223–226.
- Gower, B.A.; Fowler, L.A. Obesity in African-Americans: The role of physiology. J. Intern. Med. 2020, 288, 295–304.
- Mogensen, M.; Sahlin, K.; Fernström, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Højlund, K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2007, 56, 1592–1599.
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.P.; Hesselink, M.K.C.; et al. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 2008, 57, 2943–2949.
- Muoio, D.M. Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell 2014, 159, 1253–1262.
- Berk, E.S.; Kovera, A.J.; Boozer, C.N.; Pi-Sunyer, F.X.; Albu, J.B. Metabolic inflexibility in substrate use is present in African-American but not Caucasian healthy, premenopausal, nondiabetic women. J. Clin. Endocrinol. Metab. 2006, 91, 4099–4106.
- Hickner, R.C.; Privette, J.; McIver, K.; Barakat; Bara, H. Fatty acid oxidation in African-American and Caucasian women during physical activity. J. Appl. Physiol. 2001, 90, 2319–2324.
- Cortright, R.N.; Sandhoff, K.M.; Basilio, J.L.; Berggren, J.R.; Hickner, R.C.; Hulver, M.W.; Dohm, G.L.; Houmard, J.A. Skeletal muscle fat oxidation is increased in African-American and white women after 10 days of endurance exercise training. Obesity 2006, 14, 1201–1210.
- Fisher, G.; Windham, S.T.; Griffin, P.; Warren, J.L.; Gower, B.A.; Hunter, G.R. Associations of human skeletal muscle fiber type and insulin sensitivity, blood lipids, and vascular hemodynamics in a cohort of premenopausal women. Eur. J. Appl. Physiol. 2017, 117, 1413–1422.
- Hunter, G.R.; Weinsier, R.L.; McCarthy, J.P.; Larson-Meyer, D.E.; Newcomer, B.R. Hemoglobin, muscle oxidative capacity, and VO2max in African-American and Caucasian women. Med. Sci. Sports Exerc. 2001, 33, 1739–1743.
- Ama, P.F.M.; Simoneau, J.A.; Boulay, M.R.; Theriault, G.; Bouchard, C. Skeletal muscle characteristics in sedentary Black and Caucasian males. J. Appl. Physiol. 1986, 61, 1758–1761.
- Swift, D.L.; Johannsen, N.M.; Lavie, C.J.; Earnest, C.P.; Johnson, W.D.; Blair, S.N.; Church, T.S.; Newton, R.L. Racial differences in the response of cardiorespiratory fitness to aerobic exercise training in Caucasian and African American postmenopausal women. J. Appl. Physiol. 2013, 114, 1375–1382.
- Tanner, C.J.; Barakat, H.A.; Dohm, G.L.; Pories, W.J.; MacDonald, K.G.; Cunningham, P.R.G.; Swanson, M.S.; Houmard, J.A. Muscle fiber type is associated with obesity and weight loss. Am. J. Physiol.-Endocrinol. Metab. 2002, 282, E1191–E1196.
- Hasson, B.R.; Apovian, C.; Istfan, N. Racial/Ethnic Differences in Insulin Resistance and Beta Cell Function: Relationship to Racial Disparities in Type 2 Diabetes among African Americans versus Caucasians. Curr. Obes. Rep. 2015, 4, 241–249.
- Kodama, K.; Tojjar, D.; Yamada, S.; Toda, K.; Patel, C.J.; Butte, A.J. Ethnic differences in the relationship between insulin sensitivity and insulin response: A systematic review and meta-analysis. Diabetes Care 2013, 36, 1789–1796.
- Smith, L.M.; Yao-Borengasser, A.; Starks, T.; Tripputi, M.; Kern, P.A.; Rasouli, N. Insulin resistance in African-American and Caucasian women: Differences in lipotoxicity, adipokines, and gene expression in adipose tissue and muscle. J. Clin. Endocrinol. Metab. 2010, 95, 4441–4448.
- Privette, J.D.; Hickner, R.C.; MacDonald, K.G.; Pories, W.J.; Barakat, H.A. Fatty acid oxidation by skeletal muscle homogenates from morbidly obese black and white American women. Metab. Clin. Exp. 2003, 52, 735–738.
- Kim, J.-y.; Hickner, R.C.; Cortright, R.L.; Dohm, G.L.; Houmard, J.A. Lipid oxidation is reduced in obese human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 279, 1039–1044.
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, 578–591.
- Albu, J.B.; Kovera, A.J.; Allen, L.; Wainwright, M.; Berk, E.; Raja-Khan, N.; Janumala, I.; Burkey, B.; Heshka, S.; Gallagher, D. Independent association of insulin resistance with larger amounts of intermuscular adipose tissue and a greater acute insulin response to glucose in African American than in white nondiabetic women. Am. J. Clin. Nutr. 2005, 82, 1210–1217.
- Gallagher, D.; Kuznia, P.; Heshka, S.; Albu, J.; Heymsfield, S.B.; Goodpaster, B.; Visser, M.; Harris, T.B. Adipose tissue in muscle: A novel depot similar in size to visceral adipose tissue. Am. J. Clin. Nutr. 2005, 81, 903–910.
- Lawrence, J.C.; Newcomer, B.R.; Buchthal, S.D.; Sirikul, B.; Oster, R.A.; Hunter, G.R.; Gower, B.A. Relationship of intramyocellular lipid to insulin sensitivity may differ with ethnicity in healthy girls and women. Obesity 2011, 19, 43–48.
- Miljkovic-Gacic, I.; Gordon, C.L.; Goodpaster, B.H.; Bunker, C.H.; Patrick, A.L.; Kuller, L.H.; Wheeler, V.W.; Evans, R.W.; Zmuda, J.M. Adipose tissue infiltration in skeletal muscle: Age patterns and association with diabetes among men of African ancestry. Am. J. Clin. Nutr. 2008, 87, 1590–1595.
- Reed, R.M.; Nevitt, S.J.; Kemp, G.J.; Cuthbertson, D.J.; Whyte, M.B.; Goff, L.M. Ectopic fat deposition in populations of black African ancestry: A systematic review and meta-analysis. Acta Diabetol. 2021, 59, 171–187.
- Kitessa, S.M.; Abeywardena, M.Y. Lipid-induced insulin resistance in skeletal muscle: The chase for the culprit goes from total intramuscular fat to lipid intermediates, and finally to species of lipid intermediates. Nutrients 2016, 8, 466.
- Wigger, D.; Schumacher, F.; Schneider-Schaulies, S.; Kleuser, B. Sphingosine 1-phosphate metabolism and insulin signaling. Cell Signal. 2021, 82, 109959.
- Katzmarzyk, P.T.; Bray, G.A.; Greenway, F.L.; Johnson, W.D.; Newton, R.L.; Ravussin, E.; Ryan, D.H.; Smith, S.R.; Bouchard, C. Racial differences in abdominal depot-specific adiposity in white and African American adults. Am. J. Clin. Nutr. 2010, 91, 7–15.
- Tulloch-Reid, M.K.; Hanson, R.L.; Sebring, N.G.; Reynolds, J.C.; Premkumar, A.; Genovese, D.J.; Sumner, A.E. Both subcutaneous and visceral adipose tissue correlate highly with insulin resistance in african americans. Obes. Res. 2004, 12, 1352–1359.
- Buie, J.N.J.; Hammad, S.M.; Nietert, P.J.; Magwood, G.; Adams, R.J.; Bonilha, L.; Sims-Robinson, C. Differences in plasma levels of long chain and very long chain ceramides between African Americans and whites: An observational study. PLoS ONE 2019, 14, e0216213.
- Peterson, L.R.; Xanthakis, V.; Duncan, M.S.; Gross, S.; Friedrich, N.; Völzke, H.; Felix, S.B.; Jiang, H.; Sidhu, R.; Nauck, M.; et al. Ceramide remodeling and risk of cardiovascular events and mortality. J. Am. Heart Assoc. 2018, 7, e007931.
- Hilvo, M.; Salonurmi, T.; Havulinna, A.S.; Kauhanen, D.; Pedersen, E.R.; Tell, G.S.; Meyer, K.; Teeriniemi, A.M.; Laatikainen, T.; Jousilahti, P.; et al. Ceramide stearic to palmitic acid ratio predicts incident diabetes. Diabetologia 2018, 61, 1424–1434.
This entry is offline, you can click here to edit this entry!