Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Medicinal
Triple negative breast cancer (TNBC) is an urgent as well as huge medical challenge, which is associated with poor prognosis and responsiveness to chemotherapies. Since epigenetic changes are highly implicated in TNBC tumorigenesis and development, inhibitors of histone deacetylases (HDACIs) could represent a promising therapeutic strategy.
- anticancer
- benzamide
- chimeric compounds
- hydroxamate
1. Introduction
Breast cancer (BC) is the most prevalent cancer in females worldwide. In 2020, 2.3 million women were diagnosed with BC and 685,000 deaths were recorded [1], which represents a public health issue. Among the BC subtypes, the hormone receptor (HR) positive is the most common, followed by the human epidermal growth factor receptor 2 (HER2) positive and the so-called triple negative (TN) phenotype. Triple negative breast cancers are the most aggressive subtype, since they present high-grade invasiveness, high tendency to give rise to distant metastasis, and are poorly responsive to the neoadjuvant anthracycline- and taxane-based chemotherapies. Dramatically, the overall TNBC patients’ survival is about 1 year [2]. Therefore, the identification of new therapeutic options to treat this cancer is an urgent as well as huge medical challenge.
Epigenetic changes are highly implicated in TNBC tumorigenesis and development, and they are affected by the chromatin remodeling [3]. Histone deacetylases (HDACs, Table 1) are a family of specialized enzymes responsible for the removal of acetyl groups from the ε-amino group of the proteinogenic histone lysine residues, leading to a compressed chromatin structure and to the consequent gene transcription suppression [4]. Based on the homology with yeast deacetylases, cellular localization, size, and number of active sites, they can be divided into four classes, i.e., Class-I (HDAC isoforms 1, 2, 3, and 8); Class-IIa (HDAC isoforms 4, 5, 7, and 9); Class-IIb (HDAC isoforms 6 and 10); and Class-IV (HDAC isoforms 11). On the other hand, histone acetyltransferases (HATs) are responsible for the inverse modification, i.e., they catalyze the lysine acetylation, leading to chromatin relaxation and active gene transcription [5]. Considering their crucial role in gene transcription and translation, and their overexpression in a variety of human cancers, HDACs have received considerable attention from researchers as therapeutic targets to manage cancer initiation and progression [6]. Different HDAC inhibitors (HDACIs) have been approved by the Food and Drug Administration (FDA), particularly against hematological diseases, such as Vorinostat, Romidepsin, Belinostat, and Panobinostat (Figure 1). Moreover, HDACIs have been investigated for the treatment of solid tumors, including TNBC, although clinical trials involving single agents provided poor results. Nevertheless, recent studies emphasize the high potential impact for HDACIs in controlling TNBC, since these agents can regulate the tumor angiogenesis and metastasis [7,8].
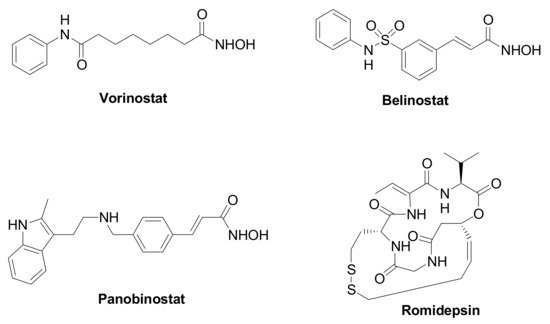
Figure 1. Chemical structure of FDA approved HDAC inhibitors.
2. HDAC Inhibitors and their Mode of Action against TNBC
2.1. Pharmacophoric Model of HDAC Inhibitors
A pharmacophoric model has been established for HDAC inhibitors based on the common structure of the different 11 zinc-dependent isoforms of the HDAC family. The deacetylase domain of these enzymes shows a surface cavity, which is connected to the inner active site by means of an intermediate channel, where the four carbons of the lysine side chain of the natural substrate are accommodated. The active site contains a Zn2+ ion, which binds to the histone N-ε acetylated lysine, with the subsequent attack to the N-acetyl group by a water molecule and the production of an N-3 free lysine and acetic acid [9]. In some HDAC isoforms, there is an additional hydrophobic cavity referred to as the “foot pocket”, where acyl side chains longer than the acetyl are accommodated [10]. Based on this enzyme architecture, HDACIs are typically endowed with a zinc binding group (ZBG) and an aromatic tail occupying the entrance area (CAP group), connected by a central lipophilic linker (Figure 2). According to the ZBG, HDAC inhibitors are classified in four classes: carboxylic acids, thiols, benzamides, and hydroxamic acids (Figure 2).
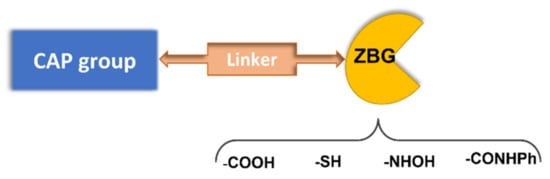
Figure 2. Pharmacophoric model for HDAC inhibitors.
The first generation of compounds obtained based on this pharmacophoric model, not exploiting the differences among the HDAC isoforms, resulted in pan-HDAC inhibitors that are often associated with numerous side effects. Indeed, the biological consequences of targeting multiple HDACs can be unpredictable, given the pleiotropic activities of these enzymes. Thanks to the increasing availability of crystallographic data and comprehensive SAR studies, significant advances in the research of HDACs isoform-selective inhibitors have been carried out [11,12]. The major differences among the HDACs are found at the entry of the active site and in the channel length and size. Moreover, the presence or the absence of the “foot-pocket” is another important difference among these metallo-enzymes, which could be exploited in the design of selective agents. For example, Entinostat and Tucidinostat are benzamide-based Class-I selective HDAC inhibitors (Figure 3) currently under clinical evaluation in both the US and Europe for the treatment of both cancer and nononcological diseases [13,14]. Ricolinostat and Citarinostat (Figure 3) are hydroxamates, which are quite selective toward the Class-II HDAC6 (HDAC1/HDAC6 selectivity index = 13 and 12, respectively) and are under evaluation against different malignancies [15,16]. In general, new and more selective HDAC6 inhibitors have been disclosed as useful tools to counteract tumor progression [17,18], although it was also reported that the selective inhibition of HDAC6 is not necessarily related to an anticancer activity, as observed for Tubathian A (Figure 3) and related compounds [19]. Therefore, increasing attention is required for the evaluation of HDAC inhibitors as anticancer drugs, especially when used as single agents. In recent years, several researches in the field of HDAC inhibitors have pointed to the hydroxamic acid and benzamide type scaffolds that selectively target HDAC. Currently, hydroxamates hold clinical success with respect to benzamides, with three FDA approved molecules (Vorinostat, Belinostat, and Panobinostat), while only Tucidinostat has been approved by the Chinese FDA to treat patients with recurrent or refractory peripheral T-cell lymphoma [20].
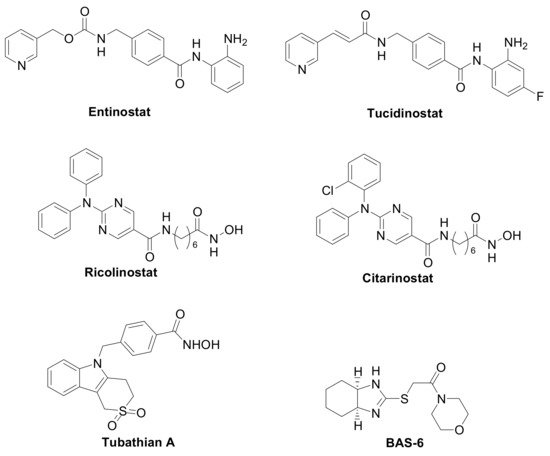
Figure 3. Class-selective HDAC inhibitors.
2.2. HDACIs Biological Action against TNBC Progression
Both unselective and selective HDACIs have been investigated against TNBC. Additionally, these studies have disclosed the different pathways influenced by HDACIs that trigger the observed antitumoral effects (Table 2).
Table 2. Summary of the main biological effects of HDACIs in TNBC cells.
| HDACIs | Action | Ref. |
|---|---|---|
| Pan-HDACIs (Vorinostat, Panobinostat) and Class-I Selective (Entinostat) | Antiproliferative, pro-apoptotic, and immunomodulatory by:
|
[7,21,23,24,28,29,30,31] |
| Class-I HDAC Selective (Entinostat) |
ER− to ER+ conversion; compromise vasculogenic mimicry data | [8,25] |
| HDAC6 Selective (BAS-6) | Alteration of the glycolytic metabolism | [27] |
In general, the pan-HDAC inhibitors Vorinostat and Panobinostat act alone as antiproliferative and pro-apoptotic agents against TNBC cell lines, such as MDA-MB-231, 4T1 and BT-549, by the modulation of multiple pathways and factors. The following factors can be mentioned: the activation of the epithelial-mesenchymal transition (EMT) phenotype and the downregulation of the growth factor FOXA1 [21], the impairment of the homologous recombination repair pathway of DNA [22], the upregulation of tumor suppressor factors p21 and p27, and the downregulation of the survival protein Bcl-2 [23]. Moreover, Vorinostat has the ability to impair BC cell migration and invasion via the inhibition of matrix metallo-proteinase 9 (MMP9) activity [24], both alone and in combination with ionizing radiation.
Interestingly, the Class-I selective HDACI Entinostat, but not pan HDACIs, had the ability to convert ER-negative tumors to ER-positive tumors, sensitizing TNBC to letrozole treatment [25,26]. Moreover, Entinostat demonstrated the ability to upregulate genes involved in the development of the vasculogenic mimicry, i.e., a tumor blood supply system independent from angiogenesis [8]. In addition, a Class-II selective inhibitor directed against the HDAC6, the BAS-6 (Figure 3), selectively kills apoptotic-resistant TNBC cells inducing their glycolytic metabolism alteration [27].
To date, the outcomes obtained in the TNBC management by HDACIs point to the significant potential of these compounds in controlling cancer proliferation and chemo-resistance.
This entry is adapted from the peer-reviewed paper 10.3390/ph15060667
This entry is offline, you can click here to edit this entry!