Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Insulin-like growth factor two (IGF-II) is a key protein regulating growth, particularly during normal fetal development, but it is also often dysregulated during tumorigenesis. IGF-II belongs to a larger system including several different regulatory factors and generally referred to as the “IGF system” that we will briefly present. IGF-I and IGF-II are the main ligands of the type 1 IGF receptor (IGF-1R).
- IGF2
- growth
- Parental imprinting
1. Main Characteristics, Linear Organization
In 2019, Baral et al. unraveled the complexity of the genomic and transcriptional organization of the IGF2/Igf2 locus, by using human or mouse DNA segments as queries in genome analyses, and RNA sequencing libraries (complete review in [Baral, K.; Rotwein, P. PLoS ONE 2019, 14, e0219155.]). IGF2 (ENSG00000167244) is composed of 10 exons and five promoters, whereas its mouse counterpart is located on chromosome 7, and is composed of eight exons and four promoters.
The five human IGF2 promoters control the expression of different non-coding exons, but all transcripts include exons 8–10, which encode the IGF-II protein precursor and the 3′ untranslated RNA. Human promoters 1 and 2 (P1 and P2) are species specific, and P2 regulates two classes of IGF2 transcripts differing due to alternative splicing of exon 5 (Figure 3).
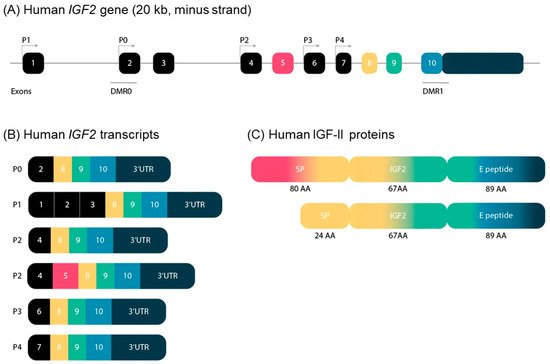
Figure 3. (A) Schematic representation of the structure of the IGF2 gene in humans. The IGF2 gene consists of 10 exons and is driven by five different promotors. The exons of the IGF2 gene are boxed. The black boxes indicate non-coding exons. The colored boxes indicate the coding exons. The turned arrows show the promotors (P) and indicate the transcription start sites. The blue lines indicate the differentially methylated regions (DMR) in the IGF2 gene. (B) Transcripts of the human IGF2 gene. IGF2 has six alternative transcripts, depending on promotors and splice sites used. (C) Human IGF-II proteins. IGF-II has two precursor proteins. Only exons 5, 8, 9 and 10 encode IGF-II proteins. Exon 5 is not included in the composition of the second precursor protein, which therefore has a smaller signal peptide. SP: signal peptide; AA: amino acids.
The human-secreted IGF-II is composed of 67 amino acids organized into four domains, the B, C, A, and D domains (listed in order from the N- to the C-terminus) [De Pagter et al FEBS Lett. 1987,214, 259–264.]. Two types of protein precursors with different presumptive N-terminal signal peptides (consisting of 24 or 80 amino acids) give rise to mature human IGF-II, depending on the inclusion or exclusion of exon 5 in the IGF2 mRNA. The E peptide of the IGF-II precursor (also named “big” IGF-II), encoded by the 3-’end mRNA, is 89 amino acids long (Figure 3). It has been involved in paraneoplastic pancreatic independent hypoglycemia [Zapf et al J. Clin. Investig. 1992, 90, 2574–2584. Rotwein et al J. Biol. Chem. 2017, 292, 9252–9261].
The 11p15.5 locus also includes H19, which is separated from IGF2 by about 80 kb (Figure 2). In both humans and mice, IGF2 and H19 are imprinted in a reciprocal manner. Moore et al. detected three Igf2 antisense transcripts relative to P0 transcription, with no open reading frames, in mice [Moore et al. Proc. Natl. Acad. Sci. USA 1997, 94, 12509–12514]. The exact role of these transcripts is unclear, and it is unknown whether their co-expression with the sense transcript (which has yet to be demonstrated), within the same cell, would influence mRNA stability.
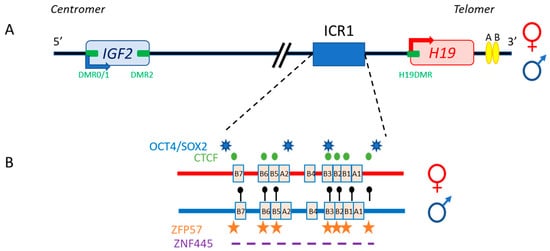
Figure 2. The human IGF2/H19 11p15.5 locus. (A) The IGF2 and H19 genes are separated by about 80 kb. IGF2 is paternally expressed (blue arrow), whereas H19 is maternally expressed (red arrow). The four DMR (green boxes) and enhancers (yellow ellipses) are represented. (B) H19/IGF2:IG-DMR (ICR1) in detail: OCT4/SOX2 (blue stars) and CTCF (green circles) binding sites on the maternal allele (red), and methylation sites (black lollipops), ZFP57 binding sites (orange stars) and the undefined ZNF445 binding site consensus sequence (purple dash) on the paternal allele (blue) are shown.
Different protein-binding sites are involved in regulating the expression of these two genes. The first evidence of differences in protein-binding sites and methylation status between the two alleles came from DNA hypersensitive site studies on the mouse Igf2/H19 locus [Khosla et al. Mol. Cell. Biol. 1999, 19, 2556–2566]. These studies revealed clear differences in nuclease sensitivity between the parental chromosomes, with the presence of mutually exclusive hypersensitive sites (on the maternal chromosome) and DNA methylation sites (on the paternal chromosome).
The specific parent-of-origin pattern of expression of H19 and IGF2 at 11p15.5 is controlled by the allele-specific methylation status of H19/IGF2:IG-DMR. This locus is composed of seven CTCF-binding sites (CBS1-7) located in the A and B blocks of repeated domains (Figure 2). CTCF is a highly conserved zinc-finger DNA-binding protein with multiple roles in gene regulation [Phillips et al. Cell 2009, 137, 1194–1211]. The region orthologous to ICR1 in mouse contains only four CBS at the Igf2 locus. Several studies in humans have reported methylation at all CBS in H19/IGF2:IG-DMR on the paternal allele and established a correlation between the methylation of these CBS and IGF2 expression from the paternal allele [Takai et al. Hum. Mol. Genet. 2001, 10, 2619–2626]. Our team has also demonstrated homogeneous methylation levels for all CBS in humans [Abi Habib, et al. Hum. Mol. Genet. 2014, 23, 5763–5773]. In mice, CTCF binding to the unmethylated maternal ICR is essential for imprint maintenance in somatic cells, providing protection against aberrant de novo methylation at DMR throughout the locus. Furthermore, CTCF acts as an insulator, and CTCF binding creates a small loop of silent chromatin (CTCF binding to the maternal ICR regulates its interaction with matrix attachment region (MAR)3 and DMR1 at Igf2) (Figure 4), preventing enhancers from gaining access to the Igf2 promoter [Kurukuti et al. Proc. Natl. Acad. Sci. USA 2006, 103, 10684–10689]. H19/IGF2:IG-DMR methylation on the paternal allele abolishes CTCF binding and ICR-mediated insulation, resulting in functional communication between promoters and enhancers, and an activation of Igf2 expression (see below).
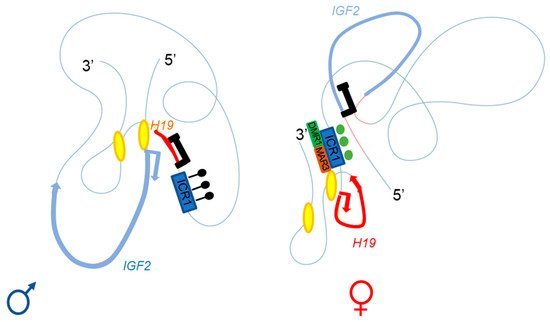
Figure 4. Adapted from Rovina et al. [Rovina, et al. Sci. Rep. 2020, 10, 8275]. Three-dimensional representation of the IGF2-H19 locus on the paternal (left) and maternal (right) chromosomes. The ICR1 of the paternal allele is methylated (black lollipops), with enhancers A and B (yellow ellipses) close to the IGF2 promoter; this conformation allows IGF2 expression and H19 repression. The CTCF of the maternal allele can bind the unmethylated ICR1 (green circles), regulating the interaction with DMR1 (green box) and the matrix attachment region (MAR)3 (brown box); the A and B (yellow ellipses) enhancers are close to the H19 promoter. This conformation allows H19 expression and IGF2 repression.
In addition to CBS, the human IGF2/H19 domain contains four binding sites for the pluripotency factors OCT4 and SOX2 [Demars et al. Hum. Mol. Genet. 2010, 19, 803–814]. There is evidence to suggest that the function of CTCF is modulated by the binding of OCT4/SOX2 to neighboring areas of DNA [Weth et al. Biochem. Cell Biol. 2011, 89, 459–468]. These factors play a role in maintaining or establishing the unmethylated status of the maternal allele. This hypothesis is strongly supported by in vitro experiments and by studies in transgenic mouse models showing that the OCT4/SOX2 binding sites in the maternal H19/IGF2:IG-DMR are essential for the full protection of DNA methylation during the establishment or maintenance phases [Zimmerman et al. PLoS ONE 2013, 8, e81962. Sakaguchi, et al. Hum. Mol. Genet. 2013, 22, 4627–4637]. Indeed, in humans, mutations of these OCT4/SOX2 binding sites in the maternal allele lead to hypermethylation of the CBS, followed by an increase in IGF2 expression leading to Beckwith–Wiedemann syndrome [Abi Habib, et al. Hum. Mutat. 2017, 38, 105–111]. Two other factors, ZFP57 and ZNF445, protect ICR from DNA demethylation after fertilization. ZNF445 seems to be sufficient on its own in humans, whereas ZFP57 and ZNF445 cooperate in rodents (Figure 2) [Quenneville et al. Mol. Cell 2011, 44, 361–372, Takahashi et al.Genes Dev. 2019, 33, 49–54]. The IGF2/H19 domain also contains several other DMR: three within the IGF2 gene (DMR0, DMR1 and DMR2) and an additional DMR located in the H19 promoter (H19DMR), all of which are secondary DMR (somatic DMR that acquire their parent-specific DNA methylation mark in somatic diploid cells) methylated on the paternal allele [Monk et al. Hum. Mol. Genet. 2006, 15, 1259–1269] (Figure 2).
2. Three-Dimensional Organization
The regulation of IGF2 expression must be considered in three dimensions, with CTCF playing a major role in this aspect. There is experimental evidence to suggest that CTCF confers allele-specific effects on transcription via long-range chromatin interactions. The generation of these data was made possible by the emergence of 3C technology [Dekker et al. Science 2002, 295, 1306–1311]. These interactions are dependent on the parental origin of the chromatin.
Series of deletions at the H19/Igf2 locus have made it possible to demonstrate the presence of several enhancers, two of which are predominantly endodermal and located 10 kb from the start site of the H19 transcript. These two enhancers target the H19 and Igf2 promoters, allowing expression of the corresponding genes (see below and Figure 2 and Figure 4) [Arney Trends Genet. 2003, 19, 17–23]. For the maternal allele, for which Igf2 expression is silent, 3C data are generally consistent with a model in which the CTCF-bound ICR contacts both the upstream DMR1 and a downstream matrix attachment region (MAR). Genetic studies have confirmed that CTCF binding to the ICR is required for both the formation of ICR-DMR1-MAR contacts and the prevention of maternal-specific enhancer-Igf2 promoter interactions. By contrast, on the paternal allele, which displays active Igf2 expression, all DMR sequences are methylated, preventing CTCF binding, and most of this region appears to be accessible, allowing more fluid contact with the enhancers [Kurukuti et al. Proc. Natl. Acad. Sci. USA 2006, 103, 10684–10689, Yoon et al. Mol. Cell. Biol. 2007, 27,3499–3510].
Recent efforts to elucidate chromatin organization at the Igf2/H19 mouse locus, based on a combination of studies of allelic CTCF binding with both high-resolution and single-cell 3D chromatin organization assays, defined topologically associated domains (TAD) [Llères et al. Genome Biol. 2019, 20, 272.]. These studies determined the dynamic structure of the imprinted Igf2-H19 domain, and showed that CTCF binding occurred at multiple sites in both alleles, exclusively in ICR1 for the maternal. Furthermore, combinations of allelic 4C-seq and DNA-FISH revealed that CTCF binding to the paternal chromosome alone was correlated with a first level of sub-TAD structure. Additional CTCF binding to the differentially methylated region on the maternal chromosome adds a further layer of sub-TAD organization. This allele-specific sub-TAD organization may, thus, provide an instructive or permissive context for the correct activation of imprinted genes during development.
In humans, a specific parent-of-origin pattern of expression through TAD generation according to CTCF binding has been described, leading to IGF2 expression or silencing [Rovina et al. Sci. Rep. 2020, 10, 8275]. As in mice, the most important proteins for TAD architecture are CTCF (which bind to the ICR) (Figure 4). Moreover, crosstalk between IGF2/H19 and the CDKN1C/KCNQ1OT1 domain (another imprinted domain located in the same chromosome region) has been detected on the basis of a higher order of chromatin folding, suggesting the involvement of a mechanism for coordinating the expression of genes with the same expression status: IGF2 and KCNQ1OT1 on the paternal allele and H19 and CDKN1C on the maternal allele [Rovina et al. Sci. Rep. 2020, 10, 8275].
3. Trans-Regulation Mechanisms
In addition to being regulated by H19/IGF2:IG-DMR methylation and the three-dimensional organization of chromatin, IGF2 can be directly regulated through the activation of its promoters by several transcription factors, including those of the oncogenic HMGA2-PLAG1 pathway [Abi Habib et al. Genet. Med. 2018, 20,250–258.]. PLAG1 (pleiomorphic adenoma gene 1) overexpression was first observed in pleiomorphic adenomas of the salivary glands, identifying PLAG1 as an oncogene [Voz et al. Cancer Res. 2000, 60, 106–113]. PLAG1 is a nuclear factor with seven zinc-finger domains that can bind IGF2 promoter P3, upregulating its transcriptional activity [Voz et al. Cancer Res. 2000, 60, 106–113]. This finding has been confirmed in various other tumors, including hepatoblastomas, lipoblastomas and leukemia (review in [Van Dyck et al. Int. J. Oncol. 2007, 30, 765–774]). Interestingly, Plag1 inactivation in mouse models results in pre- and postnatal growth retardation, despite an absence of change in Igf2 expression in embryos and pups [Hensen et al. Dev. Growth Differ. 2004, 46, 459–470].
HMGA2 (high mobility group AT-hook 2), initially named HMGI-C, is a member of the high-mobility group of proteins. Its expression is usually barely detectable in normal adult cells, but increases in cells transformed with viral oncogenes and in malignant tissues [Manfioletti et al. Nucleic Acids Res. 1991, 19, 6793–6797]. Mouse models of Hmga2 inactivation have a pygmy phenotype, with pre- and postnatal growth restriction and craniofacial abnormalities (a shortened head) [Zhou et al. Nature 1995, 376, 771–774.]. The expression levels of HMGA2 and PLAG1 are highly correlated in thyroid tumors, and HMGA2 overexpression in cellular models is associated with an increase in PLAG1 expression [Klemke et al. PLoS ONE 2014, 9, e88126].
The role of this oncogenic pathway in the control of IGF2 expression was highlighted in 2017, with the identification of additional mutations of HMGA2 and the first mutations of PLAG1 in patients referred for Silver–Russell syndrome (in addition to original mutations of IGF2). One of these PLAG1 mutations led to a downregulation of IGF2 expression in fibroblasts through a specific change in P3 promoter activity. Finally, the overexpression of HMGA2 and PLAG1 or their silencing in transfection assays result in a gain in expression or the downregulation of IGF2 expression, respectively [Abi Habib et al. Genet. Med. 2018, 20,
250–258.].
250–258.].
DIS3L2 (DIS3-like 3′-5′ exoribonuclease 2) encodes a protein involved in the processing of mRNA and small non-coding RNAs. Homozygous loss-of-function variants of DIS3L2 lead to a rare condition called Perlman syndrome. This syndrome is characterized by excessive fetal growth and an increase in the risk of Wilms’ tumor [Astuti et al. Nat. Genet. 2012,
44, 277–284]. In a mouse model, Dis3l2 invalidation was associated with an overexpression of Igf2 in nephron progenitor cells that was not associated with a loss of imprinting, as Igf2 still displayed monoallelic expression. The mechanism of Igf2 overexpression in this model remains to be determined [Hunter et al. Genes Dev. 2018, 32, 903–908.].
44, 277–284]. In a mouse model, Dis3l2 invalidation was associated with an overexpression of Igf2 in nephron progenitor cells that was not associated with a loss of imprinting, as Igf2 still displayed monoallelic expression. The mechanism of Igf2 overexpression in this model remains to be determined [Hunter et al. Genes Dev. 2018, 32, 903–908.].
Network of imprinted genes: In recent years, several studies in humans or animal models have shown that abnormalities at a given imprinted locus can impact at the expression of genes not only at the locus concerned, but also at other imprinted or non-imprinted loci [Varrault et al. Dev.Cell 2006, 11, 711–722, Gabory et al. Development 2009,136, 3413–3421, Al Adhami et al. Genome Res. 2015, 25, 353–367, Whipple et al. Mol. Cell 2020, 78, 85–95.]. This finding raised the possibility of an imprinted gene network, within which, imprinted genes are co-regulated. This pattern of regulation may partly account for the clinical overlap between imprinting disorders due to (epi)genetic defects at different imprinted loci [Eggermann et al. Genes 2021, 12, 585]. For example, a strong clinical overlap between Silver–Russell syndrome (SRS, OMIM #180860) and Temple syndrome (TS14, OMIM #616222) has been described, despite the existence of several syndrome-specific traits, including pre- and postnatal growth restriction, relative macrocephaly, feeding difficulties and a protruding forehead [Geoffron et al. J. Clin. Endocrinol. Metab. 2018, 103, 2436–2446]. IGF2 downregulation is thought to be the molecular mechanism underlying the SRS phenotype, with about 40% of SRS patients presenting hypomethylation at the H19/IGF2:IG-DMR [Wakeling et al. Nat. Rev. Endocrinol. 2017, 13, 105–124]. TS14 is mostly due to abnormalities of the imprinted 14q32.2 locus. This locus contains non-coding RNA sequences that are expressed from the maternal allele only (including the two long non-coding RNA, MEG3 and MEG8). In cases of maternal uniparental disomy of chromosome 14 or hypomethylation at the MEG3/DLK1:IG-DMR, MEG3 and MEG8 are expressed from both the paternal and maternal alleles, leading to an increase in the level of expression of these two genes. Abnormally low levels of IGF2 expression have been reported in the fibroblasts of TS14 patients, despite normal H19/IGF2:IG-DMR methylation. Furthermore, in control fibroblasts, the overexpression of MEG3 and MEG8 leads to a downregulation of IGF2. Conversely, the silencing of MEG3 and/or MEG8 in control fibroblasts leads to an upregulation of IGF2 expression. Thus, MEG3 and MEG8, which are expressed from the maternal 14q32.2 locus, regulates IGF2 expression at 11p15.5, providing support for the hypothesis of an imprinted gene network [Abi Habib et al. Sci. Adv. 2019, 5, eaau9425].
This entry is adapted from the peer-reviewed paper 10.3390/cells11121886
This entry is offline, you can click here to edit this entry!