Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The A3 adenosine receptor (A3AR) is overexpressed in pathological human cells. Piclidenoson and namodenoson are A3AR agonists with high affinity and selectivity to A3AR. Both induce apoptosis of cancer and inflammatory cells via a molecular mechanism entailing deregulation of the Wnt and the NF-κB signaling pathways.
- A3AR
- agonist
- anti-fibrosis
- anti-inflammatory
- liver cancer
- namodenoson
- non-alcoholic steatohepatitis
- piclidenoson
- psoriasis
1. Introduction
The A3 adenosine receptor (A3AR) has been positioned as a target for combatting inflammation and cancer over the last 2 decades [1][2]. The target, Gi protein-coupled A3AR, is highly expressed in inflammatory and cancer cells [2][3][4][5]. Interestingly, high A3AR expression levels are also found in peripheral blood mononuclear cells (PBMCs) of patients with inflammatory diseases and cancer, reflecting A3AR expression in pathological remote sites. Solid tumor cells, including breast, colon, small cell lung and pancreatic carcinoma, and melanoma, highly express A3AR compared to normal adjacent tissue cells. A3AR is also expressed in inflammatory cells such as synoviocytes derived from patients with rheumatoid arthritis, skin biopsies, and PBMCs from psoriasis and Crohn’s disease patients [6][7][8][9][10].
Targeting the receptor with synthetic and highly selective A3AR agonists and allosteric modulators induces anti-inflammatory and anti-cancer effects [4][11][12][13][14][15][16].
2. A3AR Agonists: Piclidenoson and Namodenoson
Piclidenoson (CF101, known generically as IB-MECA; molecular weight, 510.29 Da) and namodenoson (CF102, known generically as Cl-IB-MECA; molecular weight, 544.73 Da) are small, water-insoluble, orally bioavailable A3AR agonists. Both drugs are nucleoside derivatives, and their half-life time is 9 and 12 h, respectively. Their chemical structure is depicted in Figure 1.
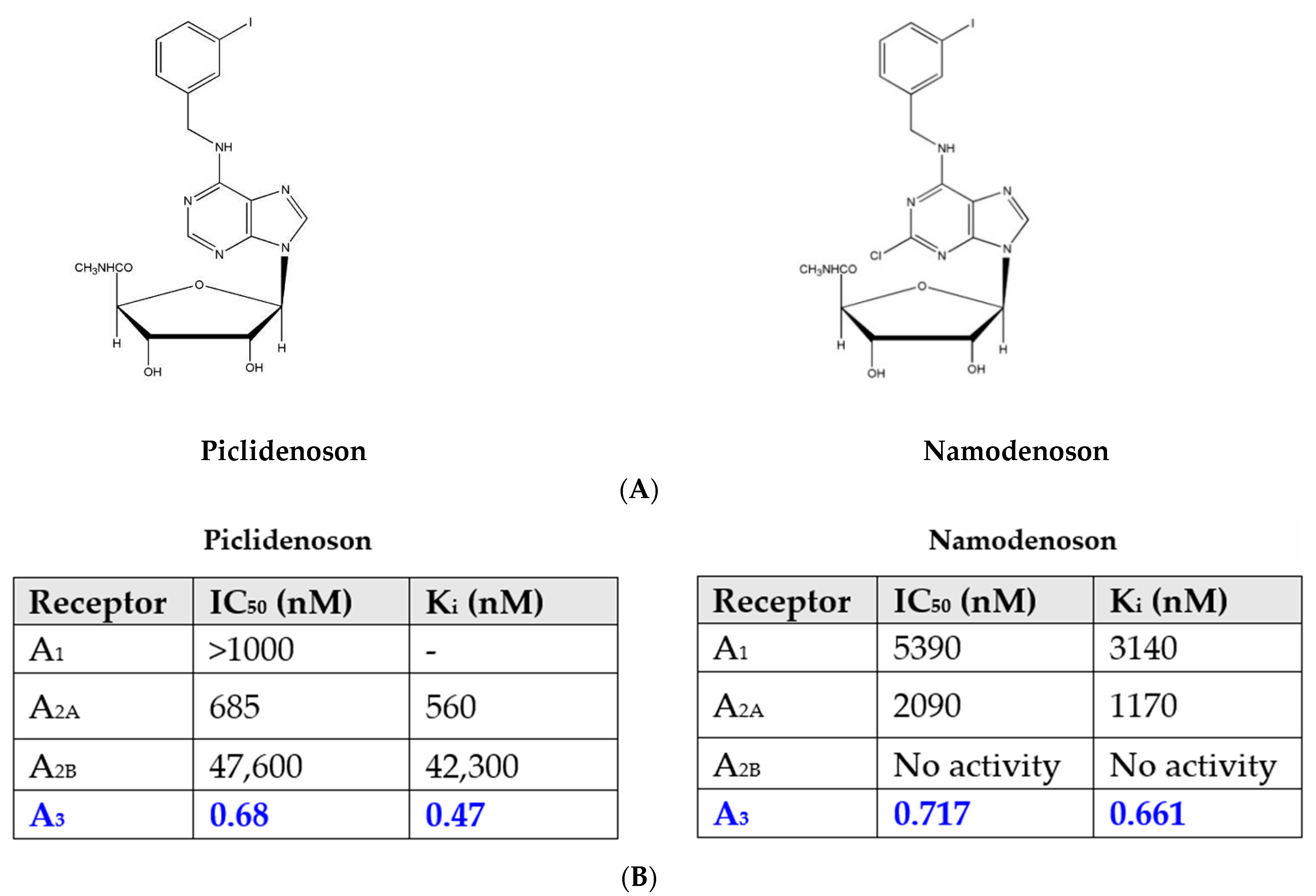
Figure 1. (A) Chemical structure of piclidenoson and namodenoson. (B) Affinity values of piclidenoson and namodenoson to the different adenosine receptors.
2.1. Piclidenoson and Namodenoson: Pharmacological Activity
The activity of piclidenoson as an orally administered, anti-inflammatory and anti-cancer agent has been tested in several experimental models of inflammatory bowel disease, arthritis, osteoarthritis, uveitis, and non-alcoholic steatohepatitis (NASH) and cytokine release syndrome [16][17][18][19][20][21][22]. In all models, oral administration of piclidenoson induced a robust reduction in the inflammatory response as measured by clinical and histological disease scores. The anti-cancer activity was demonstrated in animal models of prostate, melanoma, colon, hepatocellular, and breast carcinoma [11][12][14][23][24].
In addition, A3AR agonists mediate a differential effect on pathological and normal cells. Thus, piclidenoson and namodenoson induce apoptosis in inflammatory and cancer cells, whereas normal cells are refractory to the effect of these drugs or are affected positively by the drug [25]. In cancer cells, where there is an over-expression of the receptor, treatment with an A3AR agonist leads to inhibition of regulatory proteins such as NF-κB, followed by tumor growth inhibition [23][24]. In normal bone marrow-derived cells, upon binding of the agonist, an up-regulation of NF-κB with a subsequent increase in the level of the cytokine granulocyte colony-stimulating factor (G-CSF) takes place and induces the proliferation of normal progenitor cells with an increase in the number of white blood cells and neutrophils, yielding a chemoprotective effect [26][27]. The most prominent example involves the anti-cancer and anti-inflammatory effect of namodenoson on pathological liver cells alongside its hepatoprotective effects on normal liver cells. This differential effect attributes to the good safety profile of the A3AR agonists [28][29].
2.2. Piclidenoson and Namodenoson: Mechanism of Action
The robust anti-inflammatory and anti-cancer effects of piclidenoson and namodenoson are mediated via their ability to inhibit the production of inflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-12, interferon-ɣ, IL-17, and IL-23 [22][30][31][32]. In addition, A3AR modulation of key signaling proteins, such as PI3K, GSK-3β, PKA, PKB/Akt, IKK, and β-catenin, was shown to lead to deregulation of the NF-κB and the Wnt/β-catenin signaling pathways. This chain of events eventually induces apoptosis of inflammatory and cancer cells, resulting in anti-inflammatory and anti-cancer effects (Figure 2) [23][33][34][35][36]. These mechanistic studies laid the foundation for the clinical development of piclidenoson and namodenoson.
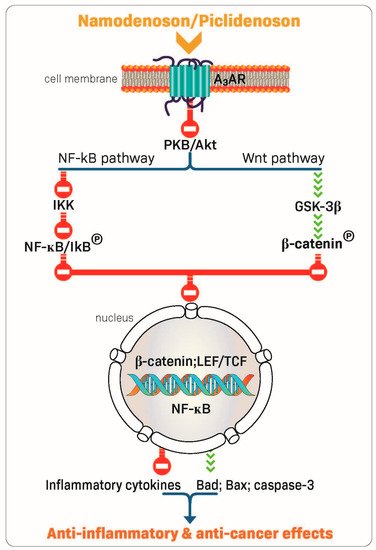
Figure 2. Mechanism of action of piclidenoson and namodenoson.
3. Piclidenoson: Clinical Development
3.1. Piclidenoson in Rheumatoid Arthritis (RA)
RA is a chronic, progressive, and disabling autoimmune disease characterized by inflammation of the joints, destruction of cartilage, and erosion of the bone. RA can make performing activities of daily living challenging and can also impact the ability to fulfill social roles and obligations. The treatments recommended by both the American College of Rheumatology (ACR) and the European Alliance of Associations for Rheumatology (EULAR) include disease-modifying antirheumatic drugs (DMARDs), such as conventional synthetic DMARDs (e.g., methotrexate (MTX)), biologic DMARDs (e.g., TNF-α inhibitors), and targeted synthetic DMARDs (e.g., JAK inhibitors) as well as glucocorticoids. Notably, these drugs are associated with adverse events, and therefore, the need exists for a new approach in anti-RA therapy [37].
The definition of response to a given drug by the ACR is scored as a percentage improvement, comparing disease activity at two discrete time points (usually a baseline vs. end of treatment). ACR20 is ≥20% improvement; ACR50 is ≥50% improvement; and ACR70 is ≥70% improvement. The ACR response criteria measures improvement in tender or swollen joint counts and improvement in at least three of the following parameters: global patient assessment of disease activity, global physician assessment of disease activity, patient pain scale, disability/functional questionnaire (Health Assessment Questionnaire Disability Index) and acute phase reactant (erythrocyte sedimentation rate [ESR] or C-reactive protein) [38]. At the same time, the definition of EULAR recommends low disease activity (LDA) as a response criterion [39].
3.1.1. Piclidenoson in RA: Phase IIa Study
In the first clinical study in RA, patients were randomized to receive piclidenoson in one of 3 doses: 0.1 mg, 1 mg, or 4 mg. The study was double-blinded with respect to the piclidenoson dose group. Piclidenoson was given as a monotherapy and administered twice daily for 12 weeks. The primary endpoint was the percentages of RA patients who met the ACR20, ACR50, and ACR70, criteria. The highest response rate was observed with 1.0 mg piclidenoson, and lower responses were found with 0.1 and 4.0 mg doses. At 12 weeks, 55.6%, 33.3%, and 11.5% of the patients receiving 1.0 mg piclidenoson achieved ACR20, ACR50 and ACR70 responses, respectively. Piclidenoson was safe and well-tolerated at all 3 doses [9].
3.1.2. Piclidenoson in RA: Phase IIb Studies
Two phase IIb studies investigated piclidenoson in combination with MTX vs. MTX alone for the treatment of RA. One study included 230 patients (NCT00556894) and the other included 252 patients (NCT00280917). In both studies, at 12 weeks, no significantly better ACR20 response rate was observed for piclidenoson plus MTX vs. MTX alone. Therefore, both studies did not reach the primary endpoint. A cross-study analysis revealed that at baseline, A3AR was highly expressed in the PBMCs derived from the phase IIa patient population. In contrast, only low receptor expression was found in the two-phase IIb patient populations, as all patients in these phase IIb studies were pre-treated with MTX. Thus, pre-treatment with MTX reduced A3AR expression and diminished the ability of piclidenoson to evoke treatment effects. It is well established that the anti-inflammatory effect of MTX is mediated via the A2A and the A3 adenosine receptors. Therefore, patients who did not respond to MTX most likely had low A2 and A3 adenosine receptor levels and thus did not respond to piclidenoson [3].
An additional phase IIb study investigated piclidenoson as a monotherapy (NCT01034306). This was a 12-week, placebo-controlled study involving 79 patients in 2 arms (twice-daily monotherapy dose of 1 mg piclidenoson vs. placebo) [3]. The study met the primary efficacy endpoint and showed statistically significant improvement vs. placebo in reducing signs and symptoms of RA, as measured by the ACR20 response rates. The ACR20 response rate in the piclidenoson arm was 49% vs. 25% in the placebo arm (p = 0.035). The ACR50 response rates were 19% and 9%, respectively, and the ACR70 response rates were 11% and 3%, respectively. The study was not designed to show statistical significance in the ACR50 and ACR70 rates. The effect of piclidenoson was linear and cumulative during the study period [40], corroborating the effect found in the earlier RA as well as psoriasis phase II studies and showing that it takes time for the drug to induce its anti-inflammatory effect.
Notably, a subgroup analysis of 16 treatment-naïve patients (8 in the piclidenoson group and 8 in the placebo group) with no prior systemic therapy showed a dramatic increase in the response for piclidenoson vs. placebo. In these patients, at 12 weeks, the ACR20 rate was 75% in the piclidenoson arm vs. 37.5% in the placebo arm. The effect of piclidenoson in treatment-naïve patients was even more pronounced for ACR50 and ACR70. ACR50 rates were 50% and 10%, respectively, and ACR70 rates were 50% and 0%, respectively. Researchers hypothesize that in treatment-naïve patients, A3AR is highly expressed.
Piclidenoson was very well-tolerated with no evidence of immunosuppression. In the piclidenoson plus MTX combination studies, no single adverse event was reported in more than one patient receiving piclidenoson, and in the monotherapy study, no severe treatment-emergent adverse events (TEAEs) were reported [40].
3.2. Piclidenoson in Psoriasis
Psoriasis is a chronic inflammatory systemic disease of the skin affecting 2–3% of the world population. It is characterized by hyperproliferation of keratinocytes and increased pro-inflammatory cytokines and chemokines in the psoriatic skin lesion, which attracts immune cells. The ability of keratinocytes to resist apoptosis plays an important role in disease pathogenesis. Furthermore, keratinocytes produce inflammatory cytokines such as TNF-α, IL-23, and IL-17 together with infiltrated immune cells, which trigger the vicious cycles of the disease [41]. Preclinical pharmacology studies demonstrated that piclidenoson inhibited the proliferation of human keratinocytes (HaCat cells). High A3AR expression levels were found in a skin biopsy and PBMCs from psoriasis patients compared to healthy subjects. Piclidenoson inhibited the proliferation of HaCat cells through deregulation of the NF-κB signaling pathway, leading to a decrease in IL-17 and IL-23 expression levels. This effect was counteracted by the specific antagonist MRS 1523. A3AR overexpression in the skin and PBMCs of psoriasis patients may be used as a target to inhibit pathological cell proliferation and the production of IL-17 and IL-23 [23].
The efficacy criteria in psoriasis are the Psoriasis Area and Severity Index (PASI) score and Physician’s Global Assessment (PGA) score. The PASI measures the average redness, thickness, and scaliness of the lesions (each graded on a 0–4 scale), weighted by the area of involvement. The PGA can be used for extensive disease as well as localised plaques. There are two primary forms: a static form, which measures the physician’s impression of the disease at a single point, and a dynamic form, in which the physician assesses the global improvement from baseline. Because the latter requires the dubious assumption that physicians can remember the severity of psoriasis at baseline throughout the trial, the static PGA has become the standard. The latter has many variations, including 5, 6, or 7-point scoring ranging from ‘clear’ to ‘severe’ [42].
3.2.1. Piclidenoson in Psoriasis: Phase II Study
The first phase II study investigating piclidenoson in psoriasis was an exploratory randomized clinical trial in patients with moderate-to-severe plaque-type psoriasis. This multicenter, randomized, double-blind, dose-ranging, placebo-controlled study involved 75 patients. Patients were treated with piclidenoson (1, 2, or 4 mg) or placebo, administered orally twice daily for 12 weeks. Safety and change from baseline in PASI and PGA scores over 12 weeks were assessed [40]. Analysis of the mean change from baseline in PASI score at week 12 revealed a statistically significant difference between the 2 mg piclidenoson-treated group and the placebo group (p < 0.001 vs. baseline and p = 0.031 vs. placebo). The 4 mg piclidenoson dose treatment resulted in reduced improvement than the 2 mg dose, and no therapeutic effect was observed with the 1 mg piclidenoson dose (Figure 3). Furthermore, in the 2 mg piclidenoson-treated group, a progressive improvement in the mean change from baseline in the PASI score throughout the study period was observed (week 2: 1.64 ± 0.9; week 4: 3.76 ± 1.9; week 8: 6.22 ± 1.9; week 12: 8.77 ± 2.1), with a statistically significant difference from placebo at weeks 8 and 12 (p = 0.047 and p = 0.031, respectively). The percentage of patients presenting only slight or no clinical signs (PGA score 0–1) increased throughout the study period in the 2 mg piclidenoson-treated group [43].
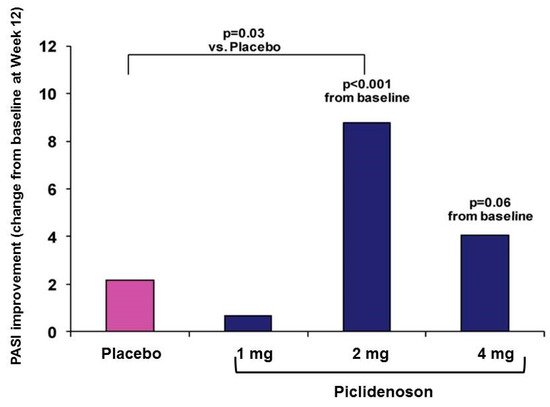
Figure 3. Change from baseline in PASI score by piclidenoson dose at week 12 [40].
Notably, this bell-shaped dose-response (e.g., highest response with the intermediate dose) is a typical response generated by piclidenoson, observed in both in vitro and in vivo studies. Such a bell-shaped response was also observed with other agonists that target Gi protein-associated cell surface receptors. This phenomenon is explained in the literature by an increase in agonist concentration that may lead to receptor desensitization, resulting in a reduced response to the given agonist.
Piclidenoson was safe and well-tolerated. No drug-related serious adverse events were reported throughout the study period [43].
4. Namodenoson: Clinical Development
4.1. Namodenson in Hepatocellular Carcinoma (HCC)
HCC is a leading cause of cancer-related death worldwide. More than 600,000 new cases are diagnosed annually worldwide [44]. Most HCC cases are associated with inflammation, usually due to hepatitis B or C virus, and occur primarily in patients with underlying cirrhosis. Thus, when selecting a treatment for HCC, both tumor burden and liver function should be considered. HCC is classified using 2 scoring systems predicting the degree of liver failure in patients with cirrhosis. The first is the Child–Pugh (CP) score, which has a strong prognostic value for HCC patients and is included in all integrated HCC staging systems, and the second is the Barcelona Clinic Liver Cancer (BCLC) system. The CP score is determined using five clinical measures, including total bilirubin, serum albumin, prothrombin time or prolongation of international normalized ratio (INR), ascites, and hepatic encephalopathy. A score of 1, 2, or 3 is given to each measure, with 3 being the most severe. The BCLC includes five prognostic factors: portal vein thrombosis, multifocal tumor, diffuse or massive disease, high alpha-fetoprotein (AFP) levels, and performance status [45].
Advanced HCC is defined as BCLC stage C, or Child–Pugh stages A-C (CPA, CPB, and CPC), and an Eastern Cooperative Oncology Group Performance Score (ECOG PS) of 1–2. Systemic therapy remains the only option for these patient populations. The current therapeutic agents approved for use in advanced HCC can be largely divided into 3 classes, beginning with tyrosine kinase inhibitors (TKI), including sorafenib, lenvatinib, cabozantinib, and regorafenib. The second group includes inhibitors of the vascular endothelial growth factor (VEGF) and the VEGF receptor (VEGFR). The third group includes immune checkpoint inhibitors against either the B7/CTLA-4 or PD-1/ L1 axis, including the anti-PD-1 antibodies, pembrolizumab, and camrelizumab, anti-PD-L1 antibodies, durvalumab, atezolizumab, and avelumab, and the anti-CTLA-4, ipilimumab. Furthermore, several treatment options are currently approved for use in the second-line setting, including nivolumab, nivolumab plus ipilimumab, pembrolizumab, cabozantinib, regorafenib, and ramucirumab [46]. However, these drugs are not given to patients defined as CPB and CPC due to their hepatotoxicity profile.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27123680
References
- Borea, P.A.; Varani, K.; Vincenzi, F.; Baraldi, P.G.; Tabrizi, M.A.; Merighi, S.; Gessi, S. The A3 adenosine receptor: History and perspectives. Pharmacol. Rev. 2015, 67, 74–102.
- Mazziotta, C.; Rotondo, J.C.; Lanzillotti, C.; Campione, G.; Martini, F.; Tognon, M. Cancer biology and molecular genetics of A3 adenosine receptor. Oncogene 2022, 41, 301–308.
- Fishman, P.; Cohen, S. The A3 adenosine receptor (A3AR): Therapeutic target and predictive biological marker in rheumatoid arthritis. Clin. Rheumatol. 2016, 35, 2359–2362.
- Madi, L.; Ochaion, A.; Rath-Wolfson, L.; Bar-Yehuda, S.; Erlanger, A.; Ohana, G.; Harish, A.; Merimski, O.; Barer, F.; Fishman, P. The A3 adenosine receptor is highly expressed in tumor versus normal cells: Potential target for tumor growth inhibition. Clin. Cancer Res. 2004, 10, 4472–4479.
- Jacobson, K.A.; Merighi, S.; Varani, K.; Borea, P.A.; Baraldi, S.; Aghazadeh Tabrizi, M.; Romagnoli, R.; Baraldi, P.G.; Ciancetta, A.; Tosh, D.K.; et al. A3 adenosine receptors as modulators of inflammation: From medicinal chemistry to therapy. Med. Res. Rev. 2018, 38, 1031–1072.
- Gessi, S.; Varani, K.; Merighi, S.; Cattabriga, E.; Avitabile, A.; Gavioli, R.; Fortini, C.; Leung, E.; Mac Lennan, S.; Borea, P.A. Expression of A3 adenosine receptors in human lymphocytes: Up-regulation in T cell activation. Mol. Pharmacol. 2004, 65, 711–719.
- Madi, L.; Cohen, S.; Ochayin, A.; Bar-Yehuda, S.; Barer, F.; Fishman, P. Overexpression of A3 adenosine receptor in peripheral blood mononuclear cells in rheumatoid arthritis: Involvement of nuclear factor-kappab in mediating receptor level. J. Rheumatol. 2007, 34, 20–26.
- Ochaion, A.; Bar-Yehuda, S.; Cohen, S.; Barer, F.; Patoka, R.; Amital, H.; Reitblat, T.; Reitblat, A.; Ophir, J.; Konfino, I.; et al. The anti-inflammatory target A3 adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and Crohn’s disease. Cell. Immunol. 2009, 258, 115–122.
- Silverman, M.H.; Strand, V.; Markovits, D.; Nahir, M.; Reitblat, T.; Molad, Y.; Rosner, I.; Rozenbaum, M.; Mader, R.; Adawi, M.; et al. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: Data from a phase II clinical trial. J. Rheumatol. 2008, 35, 41–48.
- Gessi, S.; Cattabriga, E.; Avitabile, A.; Gafa, R.; Lanza, G.; Cavazzini, L.; Bianchi, N.; Gambari, R.; Feo, C.; Liboni, A.; et al. Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Clin. Cancer Res. 2004, 10, 5895–5901.
- Fishman, P.; Bar-Yehuda, S.; Ardon, E.; Rath-Wolfson, L.; Barrer, F.; Ochaion, A.; Madi, L. Targeting the A3 adenosine receptor for cancer therapy: Inhibition of prostate carcinoma cell growth by A3AR agonist. Anticancer Res. 2003, 23, 2077–2083.
- Madi, L.; Bar-Yehuda, S.; Barer, F.; Ardon, E.; Ochaion, A.; Fishman, P. A3 adenosine receptor activation in melanoma cells: Association between receptor fate and tumor growth inhibition. J. Biol. Chem. 2003, 278, 42121–42130.
- Merimsky, O.; Bar-Yehuda, S.; Madi, L.; Fishman, P. Modulation of the A3 adenosine receptor by low agonist concentration induces antitumor and myelostimulatory effects. Drug Dev. Res. 2003, 58, 386–389.
- Ohana, G.; Bar-Yehuda, S.; Arich, A.; Madi, L.; Dreznick, Z.; Rath-Wolfson, L.; Silberman, D.; Slosman, G.; Fishman, P. Inhibition of primary colon carcinoma growth and liver metastasis by the A3 adenosine receptor agonist CF101. Br. J. Cancer 2003, 89, 1552–1558.
- Fishman, P.; Bar-Yehuda, S.; Ohana, G.; Barer, F.; Ochaion, A.; Erlanger, A.; Madi, L. An agonist to the A3 adenosine receptor inhibits colon carcinoma growth in mice via modulation of GSK-3 beta and NF-kappa b. Oncogene 2004, 23, 2465–2471.
- Bar-Yehuda, S.; Rath-Wolfson, L.; Del Valle, L.; Ochaion, A.; Cohen, S.; Patoka, R.; Zozulya, G.; Barer, F.; Atar, E.; Pina-Oviedo, S.; et al. Induction of an antiinflammatory effect and prevention of cartilage damage in rat knee osteoarthritis by CF101 treatment. Arthritis Rheum. 2009, 60, 3061–3071.
- Ren, T.; Tian, T.; Feng, X.; Ye, S.; Wang, H.; Wu, W.; Qiu, Y.; Yu, C.; He, Y.; Zeng, J.; et al. An adenosine A3 receptor agonist inhibits DSS-induced colitis in mice through modulation of the NF-kappab signaling pathway. Sci. Rep. 2015, 5, 9047.
- Baharav, E.; Bar-Yehuda, S.; Madi, L.; Silberman, D.; Rath-Wolfson, L.; Halpren, M.; Ochaion, A.; Weinberger, A.; Fishman, P. Antiinflammatory effect of a3 adenosine receptor agonists in murine autoimmune arthritis models. J. Rheumatol. 2005, 32, 469–476.
- Rath-Wolfson, L.; Bar-Yehuda, S.; Madi, L.; Ochaion, A.; Cohen, S.; Zabutti, A.; Fishman, P. IB-MECA, an A3 adenosine receptor agonist prevents bone resorption in rats with adjuvant induced arthritis. Clin. Exp. Rheumatol. 2006, 24, 400–406.
- Bar-Yehuda, S.; Luger, D.; Ochaion, A.; Cohen, S.; Patokaa, R.; Zozulya, G.; Silver, P.B.; de Morales, J.M.; Caspi, R.R.; Fishman, P. Inhibition of experimental auto-immune uveitis by the A3 adenosine receptor agonist CF101. Int. J. Mol. Med. 2011, 28, 727–731.
- Fishman, P.; Cohen, S.; Salhab, A.; Amer, J.; Itzhak, I.; Barer, F.; Safadi, R. Namodenoson anti-NAFLD/NASH activity is mediated via de-regulation of the Wnt/β-catenin pathway. Inflamm. Intest. Dis. 2019, 44, 2256–2264.
- Cohen, S.; Fishman, P. Targeting the A3 adenosine receptor to treat cytokine release syndrome in cancer immunotherapy. Drug Des. Devel. Ther. 2019, 13, 491–497.
- Bar-Yehuda, S.; Stemmer, S.M.; Madi, L.; Castel, D.; Ochaion, A.; Cohen, S.; Barer, F.; Zabutti, A.; Perez-Liz, G.; Del Valle, L.; et al. The A3 adenosine receptor agonist CF102 induces apoptosis of hepatocellular carcinoma via de-regulation of the Wnt and NF-kappab signal transduction pathways. Int. J. Oncol. 2008, 33, 287–295.
- Ledderose, C.; Hefti, M.M.; Chen, Y.; Bao, Y.; Seier, T.; Li, L.; Woehrle, T.; Zhang, J.; Junger, W.G. Adenosine arrests breast cancer cell motility by A3 receptor stimulation. Purinergic Signal. 2016, 12, 673–685.
- Fishman, P.; Bar-Yehuda, S.; Barer, F.; Madi, L.; Multani, A.F.; Pathak, S. The A3 adenosine receptor as a new target for cancer therapy and chemoprotection. Exp. Cell Res. 2001, 269, 230–236.
- Bar-Yehuda, S.; Madi, L.; Barak, D.; Mittelman, M.; Ardon, E.; Ochaion, A.; Cohn, S.; Fishman, P. Agonists to the A3 adenosine receptor induce G-CSF production via NF-κB activation: A new class of myeloprotective agents. Exp. Hematol. 2002, 30, 1390–1398.
- Cohen, S.; Stemmer, S.M.; Zozulya, G.; Ochaion, A.; Patoka, R.; Barer, F.; Bar-Yehuda, S.; Rath-Wolfson, L.; Jacobson, K.A.; Fishman, P. CF102 an A3 adenosine receptor agonist mediates anti-tumor and anti-inflammatory effects in the liver. J. Cell. Physiol. 2011, 226, 2438–2447.
- van Troostenburg, A.R.; Clark, E.V.; Carey, W.D.; Warrington, S.J.; Kerns, W.D.; Cohn, I.; Silverman, M.H.; Bar-Yehuda, S.; Fong, K.L.; Fishman, P. Tolerability, pharmacokinetics and concentration-dependent hemodynamic effects of oral CF101, an A3 adenosine receptor agonist, in healthy young men. Int. J. Clin. Pharmacol. Ther. 2004, 42, 534–542.
- Stemmer, S.M.; Benjaminov, O.; Medalia, G.; Ciuraru, N.B.; Silverman, M.H.; Bar-Yehuda, S.; Fishman, S.; Harpaz, Z.; Farbstein, M.; Cohen, S.; et al. CF102 for the treatment of hepatocellular carcinoma: A phase I/II, open-label, dose-escalation study. Oncologist 2013, 18, 25–26.
- Lee, J.Y.; Jhun, B.S.; Oh, Y.T.; Lee, J.H.; Choe, W.; Baik, H.H.; Ha, J.; Yoon, K.S.; Kim, S.S.; Kang, I. Activation of adenosine A3 receptor suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of PI 3-kinase/AKT and NF-kappab activation in murine BV2 microglial cells. Neurosci. Lett. 2006, 396, 1–6.
- Hasko, G.; Nemeth, Z.H.; Vizi, E.S.; Salzman, A.L.; Szabo, C. An agonist of adenosine A3 receptors decreases interleukin-12 and interferon-gamma production and prevents lethality in endotoxemic mice. Eur. J. Pharmacol. 1998, 358, 261–268.
- Cohen, S.; Barer, F.; Itzhak, I.; Silverman, M.H.; Fishman, P. Inhibition of IL-17 and IL -23 in human keratinocytes by the A3 adenosine receptor agonist piclidenoson. J. Immunol. Res. 2018, 2018, 2310970.
- Fishman, P.; Bar-Yehuda, S.; Madi, L.; Rath-Wolfson, L.; Ochaion, A.; Cohen, S.; Baharav, E. The PI3K-NF-kappab signal transduction pathway is involved in mediating the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis. Arthritis Res. Ther. 2006, 8, R33.
- Ochaion, A.; Bar-Yehuda, S.; Cohen, S.; Amital, H.; Jacobson, K.A.; Joshi, B.V.; Gao, Z.G.; Barer, F.; Patoka, R.; Del Valle, L.; et al. The A3 adenosine receptor agonist CF502 inhibits the PI3K, PKB/AKT and NF-kappab signaling pathway in synoviocytes from rheumatoid arthritis patients and in adjuvant-induced arthritis rats. Biochem. Pharmacol. 2008, 76, 482–494.
- Cohen, S.; Barer, F.; Bar-Yehuda, S.; AP, I.J.; Jacobson, K.A.; Fishman, P. A(3) adenosine receptor allosteric modulator induces an anti-inflammatory effect: In vivo studies and molecular mechanism of action. Mediators Inflamm. 2014, 2014, 708746.
- Fishman, P.; Bar-Yehuda, S.; Liang, B.T.; Jacobson, K.A. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov. Today 2012, 17, 359–366.
- Radu, A.F.; Bungau, S.G. Management of rheumatoid arthritis: An overview. Cells 2021, 10, 2857.
- Felson, D.T.; Lavalley, M.P. The ACR20 and defining a threshold for response in rheumatic diseases: Too much of a good thing. Arthritis Res. Ther. 2014, 16, 101–106.
- Curtis, J.R.; Trivedi, M.; Haraoui, B.; Emery, P.; Park, G.S.; Collier, D.H.; Aras, G.A.; Chung, J. Defining and characterizing sustained remission in patients with rheumatoid arthritis. Clin. Rheumatol. 2018, 37, 885–893.
- Stoilov, R.M.; Licheva, R.N.; Mihaylova, M.K.; Reitblat, T.; Dimitrov, E.A.; Shimbova, K.M.; Bhatia, G.; Pispati, A.; Gurman-Balbir, A.; Bagaria, B.R.; et al. Therapeutic effect of oral CF101 in patients with rheumatoid arthritis: A randomized, double-blind, placebo-controlled phase II study. Immunome Res. 2014, 11, 1–5.
- Zhou, X.; Chen, Y.; Cui, L.; Shi, Y.; Guo, C. Advances in the pathogenesis of psoriasis: From keratinocyte perspective. Cell Death Dis. 2022, 13, 81.
- Feldman, S.R.; Krueger, G.G. Psoriasis assessment tools in clinical trials. Ann. Rheum. Dis. 2005, 64 (Suppl. SII), ii65–ii68.
- David, M.; Akerman, L.; Ziv, M.; Kadurina, M.; Gospodinov, D.; Pavlotsky, F.; Yankova, R.; Kouzeva, V.; Ramon, M.; Silverman, M.H.; et al. Treatment of plaque-type psoriasis with oral CF101: Data from an exploratory randomized phase 2 clinical trial. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 361–367.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Subramaniam, S.; Kelley, R.B.; Venook, A.P. A review of hepatocellular carcinoma (HCC) staging systems. Chin. Clin. Oncol. 2013, 2, 33–45.
- Rimassa, L.; Worns, M.A. Navigating the new landscape of second-line treatment in advanced hepatocellular carcinoma. Liver Int. 2020, 40, 1800–1811.
This entry is offline, you can click here to edit this entry!