Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
Macrophages (Macs) are one of the major cell types of the innate immune system. They regulate inflammation and clear infection through antigen presentation, polarization, and phagocytosis. Macs release cytokines to regulate other immune cells.
- macrophage
- atherosclerosis
- polarization
1. Mac Polarization in Atherosclerotic Plaques
Local inflammatory responses in atherosclerosis activate different cells within the atherosclerotic lesion [15]. Local inflammatory responses in atherosclerosis activate different cells within the atherosclerotic lesion. Endothelial cells are activated by lipids and inflammatory mediators in the vessel wall. Modified and oxidized LDL stimulates monocytes, these engulfed monocytes then infiltrate into the arterial wall [15,16,17]. Increased levels of modified LDLs or oxidized LDLs cause the migration of a significant number of monocytes into the atherosclerotic plaque area beneath the endothelial cells in the arterial wall [16]. The inflammatory microenvironment of the lesion induces the monocytes to penetrate the arteries and differentiate into Macs. Then, Macs phagocytize the modified lipoproteins, transform into foam cells, and eventually form the atherosclerotic plaques [17]. Inflammatory Macs release pro-inflammatory cytokines and induce inflammation. Consequently, more monocytes are further recruited to the lesion area, and the accumulation of foam cells eventually leads to the formation of a necrotic core of chronic atherosclerosis [18]. Macs have an important role in the phagocytosis of necrotic cells in the plaques; pro- and anti-inflammatory Macs exacerbate or alleviate the disease, respectively [19]. The different polarization of Macs affects their proliferation and capability to recruit more monocytes which eventually alter the abundance and diversity of specific Mac cell populations in the plaques [20,21]. As shown in Figure 1, different subclasses of Macs have been identified in the plaques based on their surface markers, functions, and cytokine production [20,21,22].
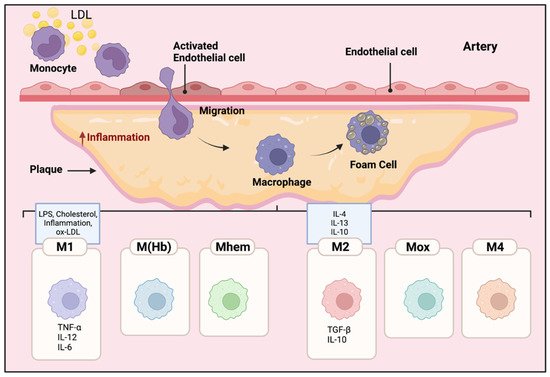
Figure 1. Mac polarization in atherosclerotic plaques. The microenvironment of the lesion affects the differentiation of monocytes to Macs. The Macs transform into foam cells and are retained in the plaques. Therefore, pro and anti-inflammatory Macs exacerbate or alleviate the disease, respectively. The polarization of Macs changes their functional phenotype in response to the signals in their microenvironment. Different subclasses of Macs have been identified in the plaques including M1, M(Hb), Mhem, M2, Mox, and M4. M1 Macs in atherosclerotic lesions can be stimulated by cholesterol crystals, LPS, pro-inflammatory cytokines, and oxidized LDL. They secrete pro-inflammatory cytokines such as TNF-α, IL-6, IL-12; in contrast, activated M2 Macs produce anti-inflammatory cytokines such as IL-10 and IL-4.
Within atherosclerotic lesions, cholesterol crystals, LPS, pro-inflammatory cytokines, and oxidized LDLs are known to induce pro-inflammatory M1 Macs [23]. Pro-inflammatory M1 Macs, normally activated through toll-like receptor (TLR-4) or nuclear factor NF kappa B (NFκB) pathways [24], secrete pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), IL-1ß, IL-6, IL-12, and IL-23 [24]. M1 Macs are the major inflammatory Mac cell population in lipid cores [25,26].
Alternatively activated M2 Macs, polarized locally or migrating from the circulating pool, produce anti-inflammatory cytokines such as IL-10 and TGF-ß, and they have a high phagocytotic capability in destroying dead cells and debris [27]. There are three subtypes of M2 macrophages. M2a Macs have been shown to have roles in wound healing, angiogenesis, and atherosclerotic lesions; M2a Macs are activated by IL-4 and IL-13 cytokines [28,29]. In contrast, M2b subtype Macs are induced by immune complexes along with IL- 1ß and LPS, they specifically express high levels of TGF-ß but different from other M2 subtypes, producing inflammatory cytokines such as IL-1ß, IL-6, and TNF-α [30,31]. Finally, M2c Macs are activated by glucocorticoids and TGF- ß; they phagocytize debris and apoptotic cells [32].
Other than M1 and M2 phenotypes, other polarized Macs in the plaques have been observed such as M(Hb), Mhem, Mox, and M4 [21,31]. M(Hb) and Mhem Macs, like M2 Macs are anti-inflammatory and produce anti-inflammatory cytokines such as IL-10, preventing the progression of plaque formation [33]. Ox-phospholipids induce Mox Macs that express IL1-ß and cyclooxygenase 2 (COX-2) and are regulated by the TLR-2 dependent metabolic pathway. Mox macrophages are counted as 30% of the total macrophages in the progressive plaques [33]. M4 Macs are categorized as pro-inflammatory and pro-atherogenic Macs in atherosclerosis [33]. M4 Macs are polarized by CXC chemokine ligand 4 (Cxcl4) associated with reduced phagocytosis, producing inflammatory cytokines and molecules such as IL-6, TNF-α, and MMP-7 [31].
These polarized Mac subtypes regulate other Mac subpopulations to modify in their microenvironment, together inducing aggravation or regression of the plaques [21,31]. It is important to note that polarized Macs have plasticity and the capability to depolarize, switching their phenotype and function based on the microenvironment [21].
2. Lipid Metabolism in Macs to Promote Anti-Inflammatory Polarization
The most efficient pathway for producing ATP in the cells is fatty acid oxidation (FAO). For example, one palmitate molecule (FA contains 16 carbons) can produce 129 ATPs [31,34,35]. Excess cholesterol causes Mac-mediated foam cell formation; Macs uptake the lipoproteins from apoptotic cells and activate signaling pathways to reduce cholesterol in the cells [35]. Through scavenger receptors such as CD36, phagocytosis, and micropinocytosis, Macs uptake modified LDLs and VLDLs, and these lipids then are catabolized in the lysosome into FA and cholesterols [36]. In the endoplasmic reticulum (ER), free cholesterols are esterified and form cholesterol fatty acid esters. If not degraded or cleared, the lipid molecules accumulate in the cytosol as lipid droplets and shift the Mac towards foam cell formation [37]. Alternative pathways exist to export free cholesterols through the cell membrane [38]. Excessive cholesterol accumulation leads to increased expression of transcription factors such as retinoid x receptor (RXR) and liver x receptor (LXR) that upregulate the ABCG1 and ABCA1 expression [39]. These lipid transporters mediate the efflux of the cholesterol via intermediate pathways to form HDLs [40]. It has also been suggested that the possibility of aqueous diffusion and facilitated diffusion pathways are involved in cholesterol transport [41].
Degradation of the lipids to fatty acids and free cholesterol in the lysosome takes place through acid lipase enzymes in a process called lipolysis [42]. After efflux, cholesterol associates with LDLs, and the fatty acids enter the FAO process [43] where they are converted into fatty acid acyl-CoA and enter the mitochondria through an enzymatic process of carnitine palmitoyltransferase 1A (CPT1A) [44]. Once in the mitochondria, carnitine is removed by the CPT-2 enzyme, and β-oxidation of FA-acyl-CoA occurs [44,45]. FAO leads to increased production of acetyl-CoA through the citric acid cycle, increasing production of NADH and FADH that consequently produce ATP through the electron transport chain in the mitochondria [46].
On the other hand, Macs need lipids for proliferation and growth [43]. Fatty acid synthesis (FAS) is a metabolic pathway that generates FA in the cytoplasm using the metabolites from the Krebs cycle, pentose phosphate pathway, and glycolysis [47]. Moreover, mTOR activation induces FAS through the transcription factor sterol regulatory element-binding protein (SREBP) [48], while mTOR inhibition leads to the activation of autophagy and lipophagy that together reduce lipid accumulation in Macs [49].
FAO activation is one of the main metabolic pathways in M2 Macs [31]. IL-4 stimulated M2 Macs induce FAO which is dependent on PPARs and their co-activator peroxisome proliferator-activated receptor gamma co-activator 1-b (PGC-1b) to increase mitochondrial biogenesis [50]. The dependency of M2 Macs on FAO is still controversial. Some publications showed that inhibited FAO in M2 Macs of humans and mice, beta-oxidation inhibitor etomoxir is not able to reduce M2 polarization in IL-4 polarized M2 Macs [51,52]. The results regarding M1 Macs revealed that modified LDL and free FA uptake are increased, the expression of the scavenger receptor is upregulated, and lipolysis and FAO are reduced [52].
It is not well understood how diverse Mac phenotypes and their associated metabolic pathways are coupled with their phenotypes, meet their energy demands, or affect atherosclerotic plaque stability. However, the current literature supports the notion that M1 Macs accumulate more lipids compared to M2 Macs; M1 Macs induce inflammation and have less activated FAO, while M2 Macs have more active FAO and FA consumption [53,54].
This entry is adapted from the peer-reviewed paper 10.3390/genes13050756
This entry is offline, you can click here to edit this entry!