Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Testicular germ cell tumors (GCTs) are highly curable malignancies. Excellent survival rates in patients with metastatic disease can be attributed to the exceptional sensitivity of GCTs to cisplatin-based chemotherapy. This hypersensitivity is probably related to alterations in the DNA repair of cisplatin-induced DNA damage, and an excessive apoptotic response. However, chemotherapy fails due to the development of cisplatin resistance in a proportion of patients, who are then considered “platinum-refractory”.
- germ cell tumors
- cisplatin
- chemoresistance
- mechanisms
1. Introduction
The molecular basis of cisplatin resistance in germ cell tumors (GCTs) appears to be multifactorial. The mechanisms of cisplatin resistance in general have been classified by some authors according to the sequence of processes that follow the introduction of the drug into the human body: pre-target, on-target and post-target mechanisms. Pre-target resistance includes alterations occurring before cisplatin binds to DNA in the cell, on-target resistance refers to alterations directly related to DNA-cisplatin adducts, and post-target resistance involves alterations in downstream signaling pathways of cisplatin-mediated DNA damage that lead to apoptosis [1][2][3][4]. Although the mechanisms within cancer cells are still the main subject of research, increasing evidence has demonstrated that the tumor microenvironment and immune cells are also important in the development of cisplatin resistance [3]. The possible mechanisms involved in cisplatin resistance are summarized in Figure 1.
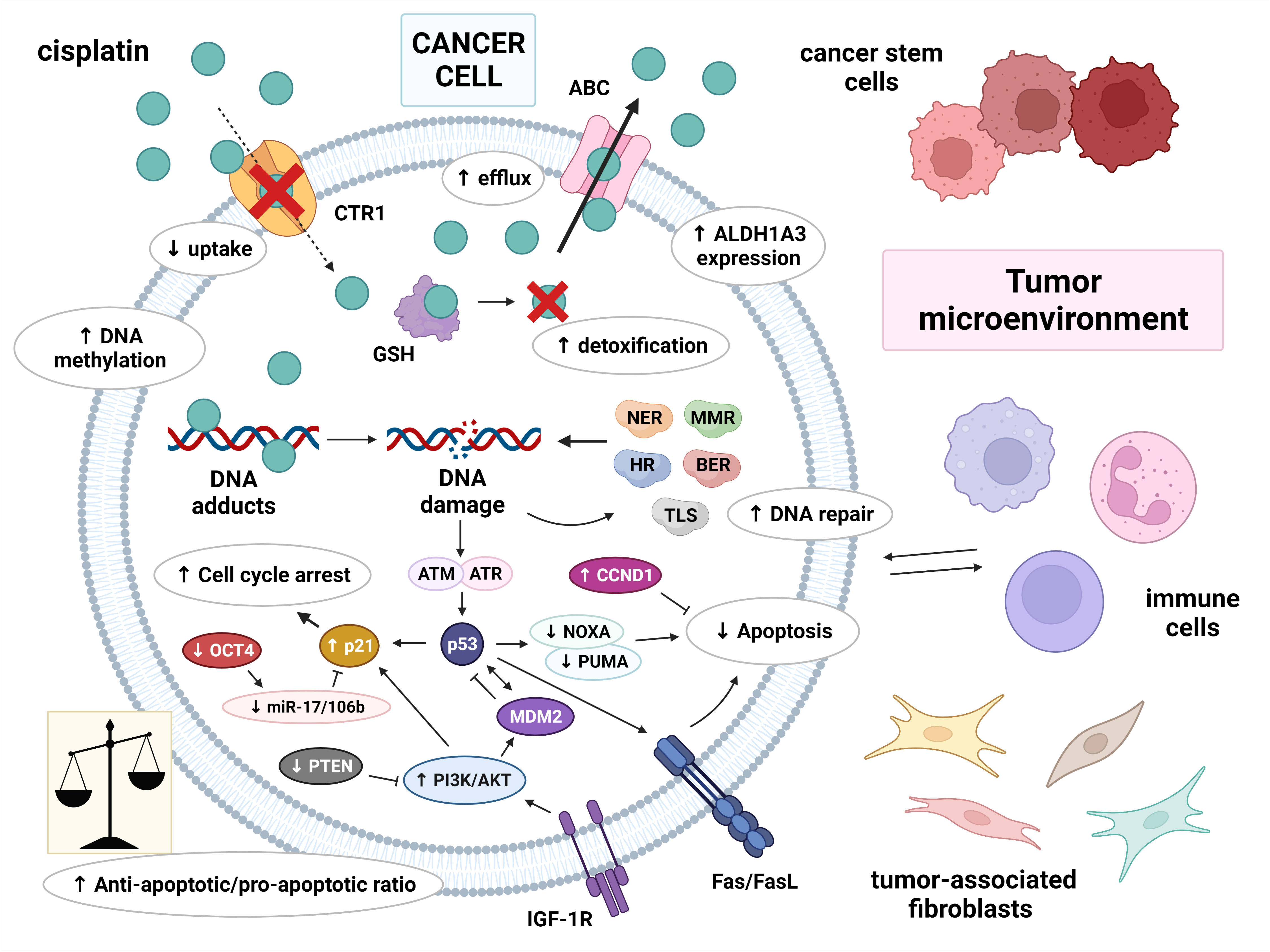
Figure 1. Multiple molecular mechanisms are responsible for cisplatin resistance in germ cell tumors. Cisplatin resistance can be classified as pre-target, on-target and post-target. Pre-target resistance includes: decreased intracellular accumulation of cisplatin (reduced uptake and increased efflux of cisplatin) and increased cisplatin detoxification by cytoplasmic scavengers. On-target resistance refers to an increased ability to repair DNA damage or an acquired ability to tolerate unrepaired DNA lesions. Post-target resistance involves: alterations in apoptosis signaling pathways with a central role of p53, decreased expression of pro-apoptotic factors, and overexpression of anti-apoptotic factors. This leads to cell cycle arrest and the inhibition of apoptosis. Other factors that contribute to cisplatin resistance are: DNA hypermethylation, increased ALDH expression, and a tumor microenvironment with immune cells playing an important role. Abbreviations: ABC = ATP-binding cassette transporter; AKT = protein kinase B (PKB); ALDH1A3 = aldehyde dehydrogenase 1A3; ATM = ataxia telangiectasia mutated; ATR = ATM and RAD3-related; BER = base excision repair; CCND1 = cyclin D1; CTR1 = copper transporter protein; FasL = Fas ligand; GSH = glutathione; HR = homologous recombination; IGF-1R = insulin-like growth factor 1 receptor; miR-17/106b = microRNA-17/106b; MDM2 = mouse double minute 2 homolog; MMR = mismatch repair; NER = nucleotide excision repair; NOXA = Phorbol-12-myristate-13-acetate-induced protein 1; OCT-4 = octamer-binding transcription factor 4; PI3K = phosphoinositide 3-kinase; PTEN = phosphatase and tensin homolog; PUMA = p53 upregulated modulator of apoptosis; TLS = translesion synthesis.
2. Pre-Target Mechanisms
Cancer cells can avoid the cytotoxic potential of cisplatin before it binds to DNA by at least two main mechanisms: decreased intracellular accumulation of cisplatin and increased cisplatin detoxification by cytoplasmic scavengers [2]. Although these mechanisms appear to be an important part of cisplatin resistance in multiple malignancies, they have not been recognized as significant sources of cisplatin resistance in GCTs. However, they may play a contributory role in GCT chemoresistance [1].
The reduced uptake of cisplatin into the cells may be mediated by the downregulation of CTR1, a transmembrane transporter for cisplatin. Studies have shown that CTR1 −/− murine embryonic fibroblasts accumulated much less cisplatin than CTR1 +/+ cells, and were more resistant to even higher concentrations of cisplatin. Furthermore, re-expression of CTR1 in the CTR1 −/− cells restored both cisplatin uptake and cytotoxicity [5][6]. Increased efflux of cisplatin is mostly regulated by the copper-transporting ATPases ATP7A and ATP7B. These transport proteins were shown to be upregulated in cisplatin-resistant cancer cell lines [7]. Studies on lung and ovarian cancer patients reported that high expression levels of ATP7A and ATP7B was correlated with a poor response to cisplatin treatment [8][9].
Resistance to cisplatin is associated with increased levels of cytoplasmic scavenger proteins, such as glutathione (GSH) and metallothionein (MT), that detoxify cisplatin through conjugation, limiting the amount of reactive cisplatin in the cell. The level of GSH appeared to be lower in GCTs compared to some other cancer types and elevated in cisplatin-resistant GCT cell lines [10]. Interestingly, one study showed high levels of a glutathione S-transferase, an enzyme responsible for the conjugation between cisplatin and GSH, in resistant teratoma [11]. Multidrug-resistance-associated proteins (MRPs) and members of ATP-binding cassette (ABC) transporters can also mediate resistance to cisplatin. MRPs are responsible for the removal of cisplatin-GSH conjugates in an ATP-dependent manner. MRP2 is mostly associated with cisplatin resistance and its high expression levels correlated with poor cisplatin response in several types of cancer [12][13].
3. On-Target Mechanisms
As mentioned before, the exceptional sensitivity of GCTs to cisplatin can be attributed to reduced capacity of repair systems towards cisplatin-induced DNA damage. Consequently, cisplatin resistance may occur due to an increased ability to repair DNA damage or an acquired ability to tolerate unrepaired DNA lesions.
One of the proposed mechanisms of cisplatin resistance may be an impaired shielding of DNA lesions from repair systems. Awuah et al. demonstrated that the knockout of HMGB4, a protein that protects cisplatin-DNA adducts from repairing through the NER system, induced resistance to cisplatin in GCT cells [14]. Another possible mechanism of GCT chemoresistance is the upregulation of NER pathway proteins, leading to a greater capacity to repair cisplatin-DNA intrastrand crosslinks. The overexpression of ERCC1 and XPF increased the repair of ICLs in GCT cell lines and rendered them more resistant to cisplatin [15]. In addition, the expression of ERCC1 and XPF proteins was shown to be higher in non-seminomas compared to seminomas and normal testis tissue. However, there was no correlation between ERCC1 and XPF expression levels on the one hand and tumor size and TNM stage on the other [16]. High ERCC1 expression was identified in resistant GCT cell lines, as well as in patients with resistant non-seminomas, but no significant association with overall survival was reported [17]. Interestingly, Cierna et al. evaluated the expression levels of NER factors in GCT patients (n = 207) and cell lines, and demonstrated the prognostic value of XPA expression on overall survival. The research showed that patients with low XPA expression had significantly better overall survival than patients with high expression (HR = 0.38, 95% CI: 0.12–1.23, p = 0.0228). Increased XPA expression was correlated with poor prognostic features in GCTs: non-seminomatous histology, high serum tumor markers, the presence of lung and non-pulmonary visceral metastases, and poor risk group according to the IGCCCG (International Germ Cell Cancer Collaborative Group) classification. In addition, XPA expression was higher in cisplatin-resistant GCT cell lines compared to sensitive ones [18].
Several studies have reported a correlation between defective MMR, microsatellite instability (MSI), BRAF mutations and cisplatin treatment failure in GCT patients [19][20][21][22]. Honecker et al. evaluated MMR proteins, MSI and BRAF mutations in 35 cisplatin-resistant GCTs compared to 100 control GCTs. Resistant tumors showed a higher incidence of MSI, as well as more BRAF mutations than controls (26% vs. 0% and 26% vs. 1%, respectively; p < 0.0001). In addition, MSI and mutated were BRAF correlated with reduced expression of MLH1 (p = 0.017 and p = 0.008, respectively) [21]. The decreased expression of MMR genes, especially MLH1 and MSH2, was also associated with a reduced cisplatin sensitivity of GCT cell lines [22]. The precise molecular mechanism in GCTs is still unclear. However, according to the accepted viewpoints, the MMR system can detect (but not repair) cisplatin-induced DNA lesions, resulting in pro-apoptotic signals. Furthermore, the lack of functional MMR proteins may lead to an increased level of translesional synthesis (TLS) and bypassing of cisplatin-DNA adducts, thus avoiding apoptosis [23][24]. TLS is a DNA damage tolerance mechanism that allows DNA replication to continue beyond cisplatin adducts. This replicative bypass is mediated by a specific group of DNA polymerases including POLH, REV1, REV3 and REV7 [25]. From the perspective of the cancer treatment of multiple malignancies, TLS increases the tolerance of cancer cells to cisplatin-induced DNA damage and allows cancer cells to survive, leading to chemoresistance [26].
4. Post-Target Mechanisms
Alterations in signal the transduction pathways that mediate apoptosis in response to DNA damage represent an essential factor in the cisplatin resistance of GCTs. Decreased expression or dysfunction of pro-apoptotic factors, as well as overexpression of anti-apoptotic factors, can lead to an altered induction of apoptosis. However, the key role of p53 mutations in GCT chemoresistance remains controversial, since mutated p53 occurs only in a subset of refractory GCTs [27][28]. Therefore, modifications of other components regulating the p53 pathway could be more important in the cisplatin resistance of GCTs. Interestingly, di Pietro et al. reported higher expression of p53, MDM2 and p21 in the intrinsic cisplatin-resistant GCT cell lines prior to cisplatin treatment compared to the cisplatin-sensitive GCT cell line and the cell subline with acquired cisplatin resistance. After cisplatin exposure, the levels of p53, MDM2 and p21 increased much more in intrinsic resistant GCT cells in comparison to sensitive GCT cells. The downregulation of p53 made cisplatin-sensitive GCT cells partially resistant to cisplatin-induced apoptosis. By contrast, p53 downregulation sensitized the intrinsically cisplatin-resistant GCT cell lines to cisplatin-induced apoptosis. These findings indicate that p53 transactivation and cisplatin-induced apoptosis in GCT cell lines depend on the cellular context. p53 appears to have a proapoptotic function in cisplatin-sensitive cells and a protective role against apoptosis in intrinsically resistant cells [29].
The PDGFR/PI3K/AKT pathway may also play a particular role in the development of cisplatin resistance. The first evidence of a dysregulation of this pathway was reported by Di Vizio et al., who showed that the tumor suppressor gene PTEN (regulating this pathway) was extensively expressed in germ cells and GCNIS, while it was practically absent from 56% of seminomas (18/32), 86% of embryonal carcinomas (19/22) and all teratomas (6/6) [30]. Later studies identified a specific role of this pathway in cisplatin resistance, in both cell lines and GCT patients. The overactivation of AKT was observed in cisplatin-resistant cells due to increased mRNA and protein levels for platelet-derived growth factor receptor b (PDGFRb) and PDGF-b ligand. Subsequently, the hyperactivation of the PDGFR/PI3K/AKT pathway resulted in an increased phosphorylation of p21 (leading to its cytoplasmic accumulation) and MDM2 (leading to the inhibition of p53-mediated apoptosis) [31][32]. Furthermore, somatic mutations in AKT1 and PIK3CA were reported exclusively in cisplatin-resistant GCTs [33]. The upregulation of insulin growth factor receptor-1 (IGF1R) expression and signaling has also been found to contribute to acquired cisplatin resistance in an in vitro non-seminoma model. IGF1R was identified as highly expressed and activated in the GCT model cell lines of non-seminoma, with the highest expression in the acquired cisplatin-resistant cell line. In addition, the silencing of IGFR1 led to the apoptosis of resistant non-seminoma cells through their re-sensitization to cisplatin [34]. The deregulation of cyclin D1 (encoded by the CCND1 gene), a cell cycle regulator, has been described as another potential cause of cisplatin resistance. The overall expression of CCND1 was higher in cisplatin-resistant cases compared to sensitive samples (p < 0.0001), with no significant differences between seminomas and non-seminomas [35].
There is increasing evidence that the cisplatin-resistant phenotype can also be maintained (if not entirely generated) by changes in signaling pathways not directly related to cisplatin, a so called “off-target” resistance. This includes the role of cellular differentiation, epigenetic mechanisms, tumor microenvironment and cancer stem cells [2].
5. The Role of Cellular Differentiation
Cellular differentiation is accompanied by epigenetic alterations that increase cisplatin resistance in GCT cells [36]. Chemoresistance is associated with more differentiated histologic subtypes of GCTs, such as teratoma. Therefore, a differentiation-inducing agent all-trans retinoic acid (ATRA) has been used to study the role of differentiation in the chemoresistance of GCT cell lines [37][38][39][40]. Gutekunst et al. demonstrated that the short-term differentiation of embryonal carcinoma cells by ATRA led to the downregulation of NOXA and PUMA, thus inhibiting apoptosis and causing a loss of cisplatin hypersensitivity [41]. The differentiation is accompanied by loss of Oct-4 expression, which has also been associated with increased resistance to cisplatin, especially due to the downregulation of NOXA and PUMA, as well as the downregulation of miR-17/-106b family members that potentiate activation of p21 [32][42][43]. Other events, such as hypoxia and cisplatin treatment, can also induce the downregulation of Oct-4 and negatively affect cisplatin sensitivity [44][45]. Interestingly, cisplatin treatment selectively depleted Oct4-positive cancer stem cells in a mouse model of metastatic GCT [46]. Taylor-Weiner et al. described the loss of pluripotency markers NANOG and Oct-4 in tumors obtained from cisplatin-resistant metastases [47]. Furthermore, in mouse models of cisplatin-sensitive and -resistant non-seminomatous GCTs, xenografts derived from cisplatin-resistant cell lines exhibited cell areas with embryonal carcinoma morphology, but no Oct-4 expression [48].
However, choriocarcinomas and yolk sac tumors, which both lack Oct-4 expression, still respond to cisplatin-based treatment. In addition, one of the first large immunohistochemical analyses of GCTs did not observe a correlation between the expression of Oct-4 and the treatment response [49]. The depletion of Oct-4 has also been shown not to alter the transactivation of p53 target genes, despite a significant decrease in cisplatin sensitivity. Therefore, it has been suggested that Oct-4 does not directly modulate p53 activity but provides a cellular environment that increases p53 proapoptotic activity by maintaining higher levels of NOXA. This indicates that NOXA levels are a central determinant of cisplatin response in GCTs, not the Oct-4 expression [42]. As already mentioned, mature teratomas, as the most differentiated subtype of GCT, that are resistant to cisplatin-based chemotherapy highly express p21 and lack Oct-4 expression. By contrast, seminomas and embryonal carcinomas that are predominantly sensitive to cisplatin hardly express p21. The expression of NOXA was also reduced in teratomas compared to seminomas and embryonal carcinomas [32][50]. The localization of p21 in teratomas was found to be primarily nuclear. However, cisplatin-resistant embryonal carcinoma cells showed high cytoplasmic p21 expression. Interestingly, the re-localization of p21 to the nucleus sensitized embryonal carcinoma cell lines to cisplatin [32].
Increasing evidence suggests that the WNT/βcatenin (WNTβ) pathway is involved in the pathogenesis and progression of GCTs. Alterations in the WNTβ pathway may contribute to cisplatin resistance [51][52][53]. The WNTβ pathway is typically activated in the early stages of embryogenesis. It also regulates the differentiation of pluripotent cells [54][55]. Furthermore, it has been associated with the process of carcinogenesis and epithelial–mesenchymal transition [56]. The translational study evaluated the clinical significance of βcatenin in GCTs and found βcatenin expression in specimens from 213 of 247 patients (86.2%). The expression in seminomas was lower compared to all subtypes of non-seminomas (all p < 0.0001). A higher expression was associated with high tumor markers (p = 0.035), primary mediastinal non-seminoma (p = 0.035) and intermediate/poor risk disease (p = 0.033) [57]. Another study reported increased expression levels of βcatenin and CCND1 in two of four cisplatin-resistant GCT cell lines and decreased levels in one embryonal carcinoma cell line. The WNT signaling inhibitor PRI-724 was not significantly effective in cell lines with increased βcatenin and CCND1 expression. However, it showed a higher pro-apoptotic effect in the cell line with decreased expression of βcatenin and CCND1, probably through the strong activation of caspase-3/7. These findings strongly suggest that the WNT/βcatenin signaling pathway is deregulated in cisplatin-resistant GCTs [58].
6. The Role of Epigenetic Mechanisms
DNA methylation as one of the main epigenetic mechanisms is closely associated with the process of tumor differentiation and seems to play a key role in the cisplatin resistance of GCTs [59][60][61][62]. Significant differences in the global DNA methylation pattern of GCTs were observed. Seminomas, which are more undifferentiated tumors that less frequently show cisplatin resistance, are hypomethylated. By contrast, more differentiated non-seminomas show a higher degree of DNA methylation. Embryonal carcinoma with an intermediate level of DNA methylation is also sensitive to cisplatin, but more often shows acquired cisplatin resistance. The highest level of DNA methylation is typical for teratoma, choriocarcinoma and yolk sac tumor, which correlates with cisplatin resistance. However, the hypermethylation of DNA was found in one case of relapsed seminoma after platinum-based chemotherapy. In addition, the demethylation of resistant seminoma cell lines resulted in an increased expression of the cell pluripotency markers NANOG and Oct-4 and a decreased resistance to cisplatin in vitro [63]. Different methylation profiles of several specific gene promoters have been reported in cisplatin-sensitive and -resistant GCTs. Promoter hypermethylation of the RASSF1A and HIC1 genes was observed in resistant non-seminomas, while sensitive non-seminomas showed hypermethylation of MGMT and RARB [64]. Martinelli et al. showed the association of CALCA hypermethylation with non-seminomas (90.5%, 19/21; p < 0.026) and cisplatin-refractory disease (47.4%, 09/19; p = 0.005). Furthermore, promoter methylation of the MGMT and CALCA genes correlated with a poor clinical outcome in GCT patients [65]. A growing number of studies have analyzed demethylating agents, their mechanism of action and its effects on GCT cell lines as well as patients. DNA methylation inhibitors are detailed in the section “Treatment approaches to overcome cisplatin resistance in germ cell tumors”.
MicroRNAs (miRNAs) are short single-stranded non-coding RNAs that modulate gene expression at the post-transcriptional level, causing inhibition of mRNA translation or its degradation. They regulate multiple biological processes and have also been linked to carcinogenesis and cancer progression, as well as chemotherapy resistance [66][67]. A study examining almost all known human micro-RNAs reported that 72 of 738 (9.8%) microRNAs were differentially expressed between cisplatin-sensitive and -resistant GCT cell lines. In addition, the miR-371-373 cluster appeared to be involved in cisplatin resistance in GCTs in vitro, since increased levels were found in two out of three resistant cell lines compared to sensitive ones. A possible mechanism might be the inhibition of the p53 pathway. The upregulation of the micro-RNA species hsa-miR-512-3p/-515/-517/-518/-525 and the downregulation of hsa-miR-99a/-100/-145 were also associated with the cisplatin-resistant phenotype in GCTs [68].
7. The Role of Tumor Microenvironment
The chemoresistance of GCTs is a complex and multifactorial phenomenon that appears to be closely related to the tumor microenvironment (TME) [69]. In general, the TME factors affecting cisplatin resistance include: physical components, such as high cell density, fluidic shear stress and extracellular matrix (ECM), which interfere with the delivery and efficacy of cisplatin, and a biological component consisting of biochemical consequences of tumor growth (hypoxia and acidity) and noncancerous cells (e.g., stromal cells, tumor-associated fibroblasts and immune cells) [3]. In addition, GCTs are infiltrated by immune cells that modulate the TME in a variety of ways, including through the secretion of cytokines. The interaction between tumor-infiltrating immune cells and cancer cells creates favorable conditions for tumor survival and growth [69][70]. A study by Siska et al. has shown that seminomas were associated with increased T cell infiltration, as well as PD-L1 expression and PD-1/PD-L1 interaction, but decreased regulatory T cells (Tregs) compared with non-seminomas. However, the advanced disease stage had different immune cell infiltration, irrespective of histological subtype. The T cell and natural killer (NK) cell populations responsible for anti-tumor immunity were decreased, while regulatory T cells (Tregs), neutrophils, mast cells and macrophages, with potentially pro-tumor immune activity, were significantly increased [71]. An intensive crosstalk between the TME and DNA damage and repair pathways has also been reported [72]. A recent study evaluated the interaction between the immune TME and endogenous DNA damage levels in GCTs by the co-cultivation of peripheral blood mononuclear cells (PBMCs) from healthy donors and GCT cell lines. PBMCs co-cultivated with cisplatin-resistant cell lines showed significantly higher DNA damage levels than PBMCs co-cultivated with sensitive cell lines. In addition, endogenous DNA damage levels above the cut-off value were associated with increased numbers of NK-cells, Tregs and CD16-positive dendritic cells [73]. Cancer cells are able to suppress anti-tumor immunity through PD-1/PD-L1 signaling in the TME. PD-L1 expression in specimens from 140 patients with GCTs was significantly higher in comparison with normal testicular tissue (p < 0.0001). Choriocarcinomas expressed the highest level of PD-L1, with declining positivity in embryonal carcinoma, teratoma, yolk sac tumor and seminoma. Furthermore, PD-L1 expression was associated with the poor prognostic features of GCTs [74]. The expression of PD-L1 was also evaluated in the tumor-infiltrating lymphocytes (TILs) of tumor samples from 240 patients with GCTs. PD-L1 expressing TILs were more frequently found in seminomas (95.9% of patients) and embryonal carcinomas (91%) compared to yolk sac tumors (60%), choriocarcinomas (54.5%) or teratomas (35.7%) (all p < 0.05). In addition, patients with high infiltration of PD-L1-positive TILs had significantly better progression-free survival (PFS) (HR = 0.17, 95% CI 0.09–0.31, p = 0.0006) and overall survival (OS) (HR = 0.08, 95% CI 0.04–0.16, p = 0.001) in contrast to patients with lower infiltration of TILs [75]. All these findings suggest a potential major role for the TME, especially immune cells, in progression, cisplatin sensitivity and resistance of GCTs.
8. The Role of Cancer Stem Cells
Cancer stem cells (CSCs) represent a subpopulation of tumor cells with cancer initiation ability, clonal long-term repopulation potential and self-renewal capability. CSCs are considered to be an origin of cancer and they can switch between stem and non-stem cell state. In addition, CSCs are resistant to conventional chemotherapy and radiation therapy [76][77][78]. Their identification is based on the expression of specific cell surface markers. CSCs have the characteristics of normal stem cells and differentiated cancer cells, and therefore they share both stemness-associated and tissue-specific markers. CD24, CD26, CD44, CD133, CD166, Ep-CAM (also called CD326 or epithelial-specific antigen) and aldehyde dehydrogenase (ALDH) are examples of CSC-specific surface markers [79].
ALDH is a NAD(P)-dependent enzyme involved in cellular detoxification and resistance to chemotherapeutic agents by oxidation of cellular aldehydes. In particular, ALDH1 family members (ALDH1A1, ALDH1A2, and ALDH1A3) are responsible for the increased self-renewal, survival and proliferation of CSCs [80]. A high expression of ALDH1 has been associated with chemoresistance and metastasis formation, and it has even been correlated with a poor clinical prognosis in several cancer types [81][82][83][84][85]. The ALDH1A3 marker was significantly overexpressed in all histological subtypes of GCTs compared to normal testicular tissue. In addition, high ALDH1A3 expression and increased ALDH activity were detected in cisplatin-resistant embryonal carcinoma cell lines. However, no association was found between ALDH1A3 expression in tumor cells and tumor primary, IGCCCG risk group, number of metastatic sites or S-stage [86].
9. Summary
The molecular mechanisms responsible for cisplatin resistance in GCTs can be classified as pre-target, on-target and post-target. Pre-target mechanisms include decreased intracellular accumulation of cisplatin - reduced cisplatin uptake by CTR1 and increased efflux of cisplatin by ABC transporters - and increased cisplatin detoxification by cytoplasmic scavengers, such as GSH. On-target mechanisms are mediated by an increased ability to repair DNA damage or an acquired ability to tolerate unrepaired DNA lesions. Post-target mechanisms involve changes in apoptosis signaling pathways, with important roles for p53, MDM2, p21 and other proteins. These processes lead to cell cycle arrest and the inhibition of apoptosis, resulting in chemoresistance. Other “off-target” factors of cisplatin resistance include cellular differentiation, epigenetic mechanisms (especially DNA hypermethylation), cancer stem cells and a tumor micro-environment with a key role for immune cells.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10050972
References
- Singh, R.; Fazal, Z.; Freemantle, S.J.; Spinella, M.J. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors. Cancer Drug Resist. 2019, 2, 580–594.
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883.
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136.
- Lobo, J.; Jeronimo, C.; Henrique, R. Cisplatin Resistance in Testicular Germ Cell Tumors: Current Challenges from Various Perspectives. Cancers 2020, 12, 1601.
- Holzer, A.K.; Manorek, G.H.; Howell, S.B. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol. Pharmacol. 2006, 70, 1390–1394.
- Larson, C.A.; Blair, B.G.; Safaei, R.; Howell, S.B. The role of the mammalian copper transporter 1 in the cellular accumulation of platinum-based drugs. Mol. Pharmacol. 2009, 75, 324–330.
- Safaei, R.; Holzer, A.K.; Katano, K.; Samimi, G.; Howell, S.B. The role of copper transporters in the development of resistance to Pt drugs. J. Inorg. Biochem. 2004, 98, 1607–1613.
- Samimi, G.; Varki, N.M.; Wilczynski, S.; Safaei, R.; Alberts, D.S.; Howell, S.B. Increase in expression of the copper transporter ATP7A during platinum drug-based treatment is associated with poor survival in ovarian cancer patients. Clin. Cancer Res. 2003, 9, 5853–5859.
- Yang, T.; Chen, M.; Chen, T.; Thakur, A. Expression of the copper transporters hCtr1, ATP7A and ATP7B is associated with the response to chemotherapy and survival time in patients with resected non-small cell lung cancer. Oncol. Lett. 2015, 10, 2584–2590.
- Masters, J.R.; Thomas, R.; Hall, A.G.; Hogarth, L.; Matheson, E.C.; Cattan, A.R.; Lohrer, H. Sensitivity of testis tumour cells to chemotherapeutic drugs: Role of detoxifying pathways. Eur. J. Cancer 1996, 32A, 1248–1253.
- Mayer, F.; Stoop, H.; Scheffer, G.L.; Scheper, R.; Oosterhuis, J.W.; Looijenga, L.H.; Bokemeyer, C. Molecular determinants of treatment response in human germ cell tumors. Clin. Cancer Res. 2003, 9, 767–773.
- Korita, P.V.; Wakai, T.; Shirai, Y.; Matsuda, Y.; Sakata, J.; Takamura, M.; Yano, M.; Sanpei, A.; Aoyagi, Y.; Hatakeyama, K.; et al. Multidrug resistance-associated protein 2 determines the efficacy of cisplatin in patients with hepatocellular carcinoma. Oncol. Rep. 2010, 23, 965–972.
- Yamasaki, M.; Makino, T.; Masuzawa, T.; Kurokawa, Y.; Miyata, H.; Takiguchi, S.; Nakajima, K.; Fujiwara, Y.; Matsuura, N.; Mori, M.; et al. Role of multidrug resistance protein 2 (MRP2) in chemoresistance and clinical outcome in oesophageal squamous cell carcinoma. Br. J. Cancer 2011, 104, 707–713.
- Awuah, S.G.; Riddell, I.A.; Lippard, S.J. Repair shielding of platinum-DNA lesions in testicular germ cell tumors by high-mobility group box protein 4 imparts cisplatin hypersensitivity. Proc. Natl. Acad. Sci. USA 2017, 114, 950–955.
- Usanova, S.; Piee-Staffa, A.; Sied, U.; Thomale, J.; Schneider, A.; Kaina, B.; Koberle, B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol. Cancer 2010, 9, 248.
- Koberle, B.; Brenner, W.; Albers, A.; Usanova, S.; Thuroff, J.W.; Kaina, B. ERCC1 and XPF expression in human testicular germ cell tumors. Oncol. Rep. 2010, 23, 223–227.
- Mendoza, J.; Martinez, J.; Hernandez, C.; Perez-Montiel, D.; Castro, C.; Fabian-Morales, E.; Santibanez, M.; Gonzalez-Barrios, R.; Diaz-Chavez, J.; Andonegui, M.A.; et al. Association between ERCC1 and XPA expression and polymorphisms and the response to cisplatin in testicular germ cell tumours. Br. J. Cancer 2013, 109, 68–75.
- Cierna, Z.; Miskovska, V.; Roska, J.; Jurkovicova, D.; Pulzova, L.B.; Sestakova, Z.; Hurbanova, L.; Machalekova, K.; Chovanec, M.; Rejlekova, K.; et al. Increased levels of XPA might be the basis of cisplatin resistance in germ cell tumours. BMC Cancer 2020, 20, 17.
- Mayer, F.; Gillis, A.J.; Dinjens, W.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Microsatellite instability of germ cell tumors is associated with resistance to systemic treatment. Cancer Res. 2002, 62, 2758–2760.
- Velasco, A.; Riquelme, E.; Schultz, M.; Wistuba, I.I.; Villarroel, L.; Pizarro, J.; Berlin, A.; Ittmann, M.; Koh, M.S.; Leach, F.S. Mismatch repair gene expression and genetic instability in testicular germ cell tumor. Cancer Biol. Ther. 2004, 3, 977–982.
- Honecker, F.; Wermann, H.; Mayer, F.; Gillis, A.J.; Stoop, H.; van Gurp, R.J.; Oechsle, K.; Steyerberg, E.; Hartmann, J.T.; Dinjens, W.N.; et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J. Clin. Oncol. 2009, 27, 2129–2136.
- Rudolph, C.; Melau, C.; Nielsen, J.E.; Vile Jensen, K.; Liu, D.; Pena-Diaz, J.; Rajpert-De Meyts, E.; Rasmussen, L.J.; Jorgensen, A. Involvement of the DNA mismatch repair system in cisplatin sensitivity of testicular germ cell tumours. Cell Oncol. 2017, 40, 341–355.
- Vaisman, A.; Varchenko, M.; Umar, A.; Kunkel, T.A.; Risinger, J.I.; Barrett, J.C.; Hamilton, T.C.; Chaney, S.G. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: Correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998, 58, 3579–3585.
- Bassett, E.; Vaisman, A.; Tropea, K.A.; McCall, C.M.; Masutani, C.; Hanaoka, F.; Chaney, S.G. Frameshifts and deletions during in vitro translesion synthesis past Pt-DNA adducts by DNA polymerases beta and eta. DNA Repair 2002, 1, 1003–1016.
- Shachar, S.; Ziv, O.; Avkin, S.; Adar, S.; Wittschieben, J.; Reissner, T.; Chaney, S.; Friedberg, E.C.; Wang, Z.; Carell, T.; et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009, 28, 383–393.
- Ler, A.A.L.; Carty, M.P. DNA Damage Tolerance Pathways in Human Cells: A Potential Therapeutic Target. Front. Oncol. 2021, 11, 822500.
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients with Advanced Germ Cell Tumors. J. Clin. Oncol. 2016, 34, 4000–4007.
- Houldsworth, J.; Xiao, H.; Murty, V.V.; Chen, W.; Ray, B.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S. Human male germ cell tumor resistance to cisplatin is linked to TP53 gene mutation. Oncogene 1998, 16, 2345–2349.
- di Pietro, A.; Koster, R.; Boersma-van Eck, W.; Dam, W.A.; Mulder, N.H.; Gietema, J.A.; de Vries, E.G.; de Jong, S. Pro- and anti-apoptotic effects of p53 in cisplatin-treated human testicular cancer are cell context-dependent. Cell Cycle 2012, 11, 4552–4562.
- Di Vizio, D.; Cito, L.; Boccia, A.; Chieffi, P.; Insabato, L.; Pettinato, G.; Motti, M.L.; Schepis, F.; D’Amico, W.; Fabiani, F.; et al. Loss of the tumor suppressor gene PTEN marks the transition from intratubular germ cell neoplasias (ITGCN) to invasive germ cell tumors. Oncogene 2005, 24, 1882–1894.
- Juliachs, M.; Munoz, C.; Moutinho, C.A.; Vidal, A.; Condom, E.; Esteller, M.; Graupera, M.; Casanovas, O.; Germa, J.R.; Villanueva, A.; et al. The PDGFRbeta-AKT pathway contributes to CDDP-acquired resistance in testicular germ cell tumors. Clin. Cancer Res. 2014, 20, 658–667.
- Koster, R.; di Pietro, A.; Timmer-Bosscha, H.; Gibcus, J.H.; van den Berg, A.; Suurmeijer, A.J.; Bischoff, R.; Gietema, J.A.; de Jong, S. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J. Clin. Investig. 2010, 120, 3594–3605.
- Feldman, D.R.; Iyer, G.; Van Alstine, L.; Patil, S.; Al-Ahmadie, H.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S.; Solit, D.B. Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant germ cell tumors. Clin. Cancer Res. 2014, 20, 3712–3720.
- Selfe, J.; Goddard, N.C.; McIntyre, A.; Taylor, K.R.; Renshaw, J.; Popov, S.D.; Thway, K.; Summersgill, B.; Huddart, R.A.; Gilbert, D.C.; et al. IGF1R signalling in testicular germ cell tumour cells impacts on cell survival and acquired cisplatin resistance. J. Pathol. 2018, 244, 242–253.
- Noel, E.E.; Yeste-Velasco, M.; Mao, X.; Perry, J.; Kudahetti, S.C.; Li, N.F.; Sharp, S.; Chaplin, T.; Xue, L.; McIntyre, A.; et al. The association of CCND1 overexpression and cisplatin resistance in testicular germ cell tumors and other cancers. Am. J. Pathol. 2010, 176, 2607–2615.
- Timmer-Bosscha, H.; de Vries, E.G.; Meijer, C.; Oosterhuis, J.W.; Mulder, N.H. Differential effects of all-trans-retinoic acid, docosahexaenoic acid, and hexadecylphosphocholine on cisplatin-induced cytotoxicity and apoptosis in a cisplantin-sensitive and resistant human embryonal carcinoma cell line. Cancer Chemother. Pharmacol. 1998, 41, 469–476.
- Skotheim, R.I.; Lind, G.E.; Monni, O.; Nesland, J.M.; Abeler, V.M.; Fossa, S.D.; Duale, N.; Brunborg, G.; Kallioniemi, O.; Andrews, P.W.; et al. Differentiation of human embryonal carcinomas in vitro and in vivo reveals expression profiles relevant to normal development. Cancer Res. 2005, 65, 5588–5598.
- Honecker, F.; Rohlfing, T.; Harder, S.; Braig, M.; Gillis, A.J.; Glaesener, S.; Barett, C.; Bokemeyer, C.; Buck, F.; Brummendorf, T.H.; et al. Proteome analysis of the effects of all-trans retinoic acid on human germ cell tumor cell lines. J. Proteom. 2014, 96, 300–313.
- Nettersheim, D.; Gillis, A.; Biermann, K.; Looijenga, L.H.; Schorle, H. The seminoma cell line TCam-2 is sensitive to HDAC inhibitor depsipeptide but tolerates various other chemotherapeutic drugs and loss of NANOG expression. Genes Chromosomes Cancer 2011, 50, 1033–1042.
- Nettersheim, D.; Arndt, I.; Sharma, R.; Riesenberg, S.; Jostes, S.; Schneider, S.; Holzel, M.; Kristiansen, G.; Schorle, H. The cancer/testis-antigen PRAME supports the pluripotency network and represses somatic and germ cell differentiation programs in seminomas. Br. J. Cancer 2016, 115, 454–464.
- Gutekunst, M.; Oren, M.; Weilbacher, A.; Dengler, M.A.; Markwardt, C.; Thomale, J.; Aulitzky, W.E.; van der Kuip, H. p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cisplatin. PLoS ONE 2011, 6, e19198.
- Gutekunst, M.; Mueller, T.; Weilbacher, A.; Dengler, M.A.; Bedke, J.; Kruck, S.; Oren, M.; Aulitzky, W.E.; van der Kuip, H. Cisplatin hypersensitivity of testicular germ cell tumors is determined by high constitutive Noxa levels mediated by Oct-4. Cancer Res. 2013, 73, 1460–1469.
- Mueller, T.; Mueller, L.P.; Luetzkendorf, J.; Voigt, W.; Simon, H.; Schmoll, H.J. Loss of Oct-3/4 expression in embryonal carcinoma cells is associated with induction of cisplatin resistance. Tumour. Biol. 2006, 27, 71–83.
- Wu, Y.C.; Ling, T.Y.; Lu, S.H.; Kuo, H.C.; Ho, H.N.; Yeh, S.D.; Shen, C.N.; Huang, Y.H. Chemotherapeutic sensitivity of testicular germ cell tumors under hypoxic conditions is negatively regulated by SENP1-controlled sumoylation of OCT4. Cancer Res. 2012, 72, 4963–4973.
- Abada, P.B.; Howell, S.B. Cisplatin induces resistance by triggering differentiation of testicular embryonal carcinoma cells. PLoS ONE 2014, 9, e87444.
- Pierpont, T.M.; Lyndaker, A.M.; Anderson, C.M.; Jin, Q.; Moore, E.S.; Roden, J.L.; Braxton, A.; Bagepalli, L.; Kataria, N.; Hu, H.Z.; et al. Chemotherapy-Induced Depletion of OCT4-Positive Cancer Stem Cells in a Mouse Model of Malignant Testicular Cancer. Cell Rep. 2017, 21, 1896–1909.
- Taylor-Weiner, A.; Zack, T.; O’Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 2016, 540, 114–118.
- Mueller, T.; Mueller, L.P.; Holzhausen, H.J.; Witthuhn, R.; Albers, P.; Schmoll, H.J. Histological evidence for the existence of germ cell tumor cells showing embryonal carcinoma morphology but lacking OCT4 expression and cisplatin sensitivity. Histochem. Cell Biol. 2010, 134, 197–204.
- Looijenga, L.H.; Stoop, H.; de Leeuw, H.P.; de Gouveia Brazao, C.A.; Gillis, A.J.; van Roozendaal, K.E.; van Zoelen, E.J.; Weber, R.F.; Wolffenbuttel, K.P.; van Dekken, H.; et al. POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors. Cancer Res. 2003, 63, 2244–2250.
- Koster, R.; van Vugt, M.A.; Timmer-Bosscha, H.; Gietema, J.A.; de Jong, S. Unravelling mechanisms of cisplatin sensitivity and resistance in testicular cancer. Expert Rev. Mol. Med. 2013, 15, e12.
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406.
- Lafin, J.T.; Bagrodia, A.; Woldu, S.; Amatruda, J.F. New insights into germ cell tumor genomics. Andrology 2019, 7, 507–515.
- Young, J.C.; Kerr, G.; Micati, D.; Nielsen, J.E.; Rajpert-De Meyts, E.; Abud, H.E.; Loveland, K.L. WNT signalling in the normal human adult testis and in male germ cell neoplasms. Hum. Reprod 2020, 35, 1991–2003.
- ten Berge, D.; Kurek, D.; Blauwkamp, T.; Koole, W.; Maas, A.; Eroglu, E.; Siu, R.K.; Nusse, R. Embryonic stem cells require Wnt proteins to prevent differentiation to epiblast stem cells. Nat. Cell Biol. 2011, 13, 1070–1075.
- Bayerl, J.; Ayyash, M.; Shani, T.; Manor, Y.S.; Gafni, O.; Massarwa, R.; Kalma, Y.; Aguilera-Castrejon, A.; Zerbib, M.; Amir, H.; et al. Principles of signaling pathway modulation for enhancing human naive pluripotency induction. Cell Stem. Cell 2021, 28, 1549–1565.
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84.
- Chovanec, M.; Cierna, Z.; Miskovska, V.; Machalekova, K.; Kalavska, K.; Rejlekova, K.; Svetlovska, D.; Macak, D.; Spanik, S.; Kajo, K.; et al. betacatenin is a marker of poor clinical characteristics and suppressed immune infiltration in testicular germ cell tumors. BMC Cancer 2018, 18, 1062.
- Schmidtova, S.; Kalavska, K.; Liskova, V.; Plava, J.; Miklikova, S.; Kucerova, L.; Matuskova, M.; Rojikova, L.; Cierna, Z.; Rogozea, A.; et al. Targeting of Deregulated Wnt/beta-Catenin Signaling by PRI-724 and LGK974 Inhibitors in Germ Cell Tumor Cell Lines. Int. J. Mol. Sci. 2021, 22, 4263.
- Buljubasic, R.; Buljubasic, M.; Bojanac, A.K.; Ulamec, M.; Vlahovic, M.; Jezek, D.; Bulic-Jakus, F.; Sincic, N. Epigenetics and testicular germ cell tumors. Gene 2018, 661, 22–33.
- Facchini, G.; Rossetti, S.; Cavaliere, C.; D’Aniello, C.; Di Franco, R.; Iovane, G.; Grimaldi, G.; Piscitelli, R.; Muto, P.; Botti, G.; et al. Exploring the molecular aspects associated with testicular germ cell tumors: A review. Oncotarget 2018, 9, 1365–1379.
- Sonnenburg, D.; Spinella, M.J.; Albany, C. Epigenetic Targeting of Platinum Resistant Testicular Cancer. Curr. Cancer Drug Targets 2016, 16, 789–795.
- Fazal, Z.; Singh, R.; Fang, F.; Bikorimana, E.; Baldwin, H.; Corbet, A.; Tomlin, M.; Yerby, C.; Adra, N.; Albany, C.; et al. Hypermethylation and global remodelling of DNA methylation is associated with acquired cisplatin resistance in testicular germ cell tumours. Epigenetics 2021, 16, 1071–1084.
- Wermann, H.; Stoop, H.; Gillis, A.J.; Honecker, F.; van Gurp, R.J.; Ammerpohl, O.; Richter, J.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Global DNA methylation in fetal human germ cells and germ cell tumours: Association with differentiation and cisplatin resistance. J. Pathol. 2010, 221, 433–442.
- Koul, S.; McKiernan, J.M.; Narayan, G.; Houldsworth, J.; Bacik, J.; Dobrzynski, D.L.; Assaad, A.M.; Mansukhani, M.; Reuter, V.E.; Bosl, G.J.; et al. Role of promoter hypermethylation in Cisplatin treatment response of male germ cell tumors. Mol. Cancer 2004, 3, 16.
- Martinelli, C.; Lengert, A.V.H.; Carcano, F.M.; Silva, E.C.A.; Brait, M.; Lopes, L.F.; Vidal, D.O. MGMT and CALCA promoter methylation are associated with poor prognosis in testicular germ cell tumor patients. Oncotarget 2017, 8, 50608–50617.
- Acunzo, M.; Romano, G.; Wernicke, D.; Croce, C.M. MicroRNA and cancer—A brief overview. Adv. Biol. Regul. 2015, 57, 1–9.
- Das, M.K.; Haugen, O.P.; Haugen, T.B. Diverse Roles and Targets of miRNA in the Pathogenesis of Testicular Germ Cell Tumour. Cancers 2022, 14, 1190.
- Port, M.; Glaesener, S.; Ruf, C.; Riecke, A.; Bokemeyer, C.; Meineke, V.; Honecker, F.; Abend, M. Micro-RNA expression in cisplatin resistant germ cell tumor cell lines. Mol. Cancer 2011, 10, 52.
- Kalavska, K.; Kucerova, L.; Schmidtova, S.; Chovanec, M.; Mego, M. Cancer Stem Cell Niche and Immune-Active Tumor Microenvironment in Testicular Germ Cell Tumors. Adv. Exp. Med. Biol. 2020, 1226, 111–121.
- Loveland, K.L.; Klein, B.; Pueschl, D.; Indumathy, S.; Bergmann, M.; Loveland, B.E.; Hedger, M.P.; Schuppe, H.C. Cytokines in Male Fertility and Reproductive Pathologies: Immunoregulation and Beyond. Front. Endocrinol. 2017, 8, 307.
- Siska, P.J.; Johnpulle, R.A.N.; Zhou, A.; Bordeaux, J.; Kim, J.Y.; Dabbas, B.; Dakappagari, N.; Rathmell, J.C.; Rathmell, W.K.; Morgans, A.K.; et al. Deep exploration of the immune infiltrate and outcome prediction in testicular cancer by quantitative multiplexed immunohistochemistry and gene expression profiling. Oncoimmunology 2017, 6, e1305535.
- Lama-Sherpa, T.D.; Shevde, L.A. An Emerging Regulatory Role for the Tumor Microenvironment in the DNA Damage Response to Double-Strand Breaks. Mol. Cancer Res. 2020, 18, 185–193.
- Kalavska, K.; Sestakova, Z.; Mlcakova, A.; Kozics, K.; Gronesova, P.; Hurbanova, L.; Miskovska, V.; Rejlekova, K.; Svetlovska, D.; Sycova-Mila, Z.; et al. Are Changes in the Percentage of Specific Leukocyte Subpopulations Associated with Endogenous DNA Damage Levels in Testicular Cancer Patients? Int. J. Mol. Sci. 2021, 22, 8281.
- Cierna, Z.; Mego, M.; Miskovska, V.; Machalekova, K.; Chovanec, M.; Svetlovska, D.; Hainova, K.; Rejlekova, K.; Macak, D.; Spanik, S.; et al. Prognostic value of programmed-death-1 receptor (PD-1) and its ligand 1 (PD-L1) in testicular germ cell tumors. Ann. Oncol. 2016, 27, 300–305.
- Chovanec, M.; Cierna, Z.; Miskovska, V.; Machalekova, K.; Svetlovska, D.; Kalavska, K.; Rejlekova, K.; Spanik, S.; Kajo, K.; Babal, P.; et al. Prognostic role of programmed-death ligand 1 (PD-L1) expressing tumor infiltrating lymphocytes in testicular germ cell tumors. Oncotarget 2017, 8, 21794–21805.
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472.
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem. Cell 2015, 16, 225–238.
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell Physiol. 2019, 234, 8381–8395.
- Dianat-Moghadam, H.; Heidarifard, M.; Jahanban-Esfahlan, R.; Panahi, Y.; Hamishehkar, H.; Pouremamali, F.; Rahbarghazi, R.; Nouri, M. Cancer stem cells-emanated therapy resistance: Implications for liposomal drug delivery systems. J. Control Release 2018, 288, 62–83.
- Bai, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Cancer stem cell in breast cancer therapeutic resistance. Cancer Treat Rev. 2018, 69, 152–163.
- Xu, S.L.; Zeng, D.Z.; Dong, W.G.; Ding, Y.Q.; Rao, J.; Duan, J.J.; Liu, Q.; Yang, J.; Zhan, N.; Liu, Y.; et al. Distinct patterns of ALDH1A1 expression predict metastasis and poor outcome of colorectal carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 2976–2986.
- Liu, Y.; Lv, D.L.; Duan, J.J.; Xu, S.L.; Zhang, J.F.; Yang, X.J.; Zhang, X.; Cui, Y.H.; Bian, X.W.; Yu, S.C. ALDH1A1 expression correlates with clinicopathologic features and poor prognosis of breast cancer patients: A systematic review and meta-analysis. BMC Cancer 2014, 14, 444.
- Li, X.; Wan, L.; Geng, J.; Wu, C.L.; Bai, X. Aldehyde dehydrogenase 1A1 possesses stem-like properties and predicts lung cancer patient outcome. J. Thorac. Oncol. 2012, 7, 1235–1245.
- Nishikawa, S.; Konno, M.; Hamabe, A.; Hasegawa, S.; Kano, Y.; Ohta, K.; Fukusumi, T.; Sakai, D.; Kudo, T.; Haraguchi, N.; et al. Aldehyde dehydrogenase high gastric cancer stem cells are resistant to chemotherapy. Int. J. Oncol. 2013, 42, 1437–1442.
- Kang, E.J.; Jung, H.; Woo, O.H.; Park, K.H.; Woo, S.U.; Yang, D.S.; Kim, A.R.; Lee, J.B.; Kim, Y.H.; Kim, J.S.; et al. Association of aldehyde dehydrogenase 1 expression and biologically aggressive features in breast cancer. Neoplasma 2014, 61, 352–362.
- Schmidtova, S.; Kalavska, K.; Gercakova, K.; Cierna, Z.; Miklikova, S.; Smolkova, B.; Buocikova, V.; Miskovska, V.; Durinikova, E.; Burikova, M.; et al. Disulfiram Overcomes Cisplatin Resistance in Human Embryonal Carcinoma Cells. Cancers 2019, 11, 1224.
This entry is offline, you can click here to edit this entry!