Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
GLP-1 receptor agonists stimulate GLP-1R to promote insulin secretion, whereas DPP4 inhibitors slow GLP-1 degradation. Both approaches are incretin-based therapies for T2D. In addition to GLP-1 analogs, small nonpeptide GLP-1RAs such as LY3502970, TT-OAD2, and PF-06882961 have been considered as possible therapeutic alternatives.
- Pseudostellaria heterophylla
- Linum usitatissimum
- Drymaria diandra
- cyclic peptides
- diabetes
- GLP-1
- DPP4
- Heterophyllin B
- Cyclolinopeptide
- Diandrine C
1. GLP-1, GLP-1R Agonists, and Type 2 Diabetes Treatment
Due to urbanization and changes in eating habits, the incidence of type 2 diabetes (T2D) has exploded. As a result, many people’s lives are greatly affected by diabetes, and the complications it causes can further harm health [1]. GLP-1 receptor agonists (GLP-1RAs) and DPP4 inhibitors are incretin-based therapies for type 2 diabetes and are critical second-line drugs for the treatment of T2D. Glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are two important incretin hormones that help maintain blood glucose homeostasis [2][3]. Of the two, GLP-1 has received more attention than GIP. GLP-1 is produced and secreted by intestinal enteroendocrine L-cells upon food consumption, and acts on pancreatic islets to stimulate insulin synthesis by activating the GLP-1 receptor (GLP-1R). GLP-1 (7–37) and GLP-1 (7–36) NH2 are their two bioactive forms. Both are rapidly degraded by Dipeptidyl peptidase 4 (DPP4) into GLP-1 (9–37) and GLP-1 (9–36) NH2 after release (t 1/2~1–2 min). The DPP4-degraded form of GLP-1 has relatively low binding affinity with the GLP-1R [2][3]. Studies have observed that patients with type 2 diabetes may not be able to maintain glucose homeostasis due to reduced GLP-1 secretion or accelerated GLP-1 metabolism [4]. Therefore, DPP4 inhibitors have been developed to prolong the effect of GLP-1, or to apply GLP-1 analogs such as exenatide and liraglutide to increase the effect on GLP-1R [5]. According to a meta-analysis, DPP4 inhibitors and GLP-1R agonists affect glucose-lowering and weight control, but GLP-1R agonists are more effective than DPP4 inhibitors [6].
Human GLP-1R is a G-protein-coupled receptor (GPCR) composed of a hydrophilic extracellular domain (ECD) and seven α-helical transmembrane domains (TMD) with 463 amino acids [7]. It is widely expressed in the lung, kidney, heart, pancreatic islets, intestines, multiple regions of the CNS, etc., and is particularly abundant in the pancreatic β-cells. The GLP-1R activation on β-cells generates a series of downstream signal amplification, including rapidly increasing cAMP levels, extracellular Ca2+ influx, β-arrestin recruitment, and ERK1/2 cascade. The cAMP-dependent activation of the PKA leads to increased insulin synthesis and release [3][8]. GLP-1R activation in the central hypothalamus has been shown to promote satiety, thereby reducing food intake and slowing gastric emptying [9]. The higher dose of liraglutide (3 mg) under the trade name Saxenda® has been approved to treat overweight and weight-related diseases [10][11]. Semaglutide is being evaluated as a weight-loss drug for obese subjects without diabetes [11]. GLP-1R agonists also have been observed to improve endothelial dysfunction that may occur in diabetic patients through blood-flow-mediated vasodilation [12]. Moreover, studies have demonstrated that GLP-1R agonists can reduce the risk of proteinuria and kidney disease progression [13]. The GLP-1R agonist is therefore considered to bring new prospects for the treatment of T2D and the prevention of its chronic complications [3][8].
Exendin-4 is a short peptide with 39 amino acids isolated from the venom of the Gila monster. It has a 53% sequence identity with human endogenous GLP-1 and was found to have an agonistic effect on GLP-1R. Exenatide (Byetta®, Bydureon®) is a synthetic version of exendin-4 and the first hypoglycemic drug classified as a GLP-1 analog [14]. Liraglutide (Victoza®) has a 97% homology with human GLP-1. In design, liraglutide conjugates fatty acids to the original GLP-1 structure. This approach strengthens its binding to serum albumin, thereby avoiding rapid degradation by DPP4 and reducing renal clearance for prolonged action. Novel semaglutide (Ozempic®) incorporates an absorption enhancer, further realizing the possibility of oral administration [8][15]. The crystal structure of the complex of exendin-4 and GLP-1R shows that except for a part of the C-terminus, exendin-4 still adopts an α-helical conformation, and its binding site is similar to that of GLP-1 [16]. When endogenous GLP-1 penetrates the core of the receptor (PDB:6X18), it interacts with Tyr205, Arg299, Thr298, Asn300, Gln234, Try152, Arg190, etc., resulting in the outward movement of TM6, ECL3, and TM7, and the inward movement of TM1 as well as the reorganization of ECL2, as shown in Figure 1. These displacements later cause the activation of GLP-1R and trigger cAMP production [3][17].
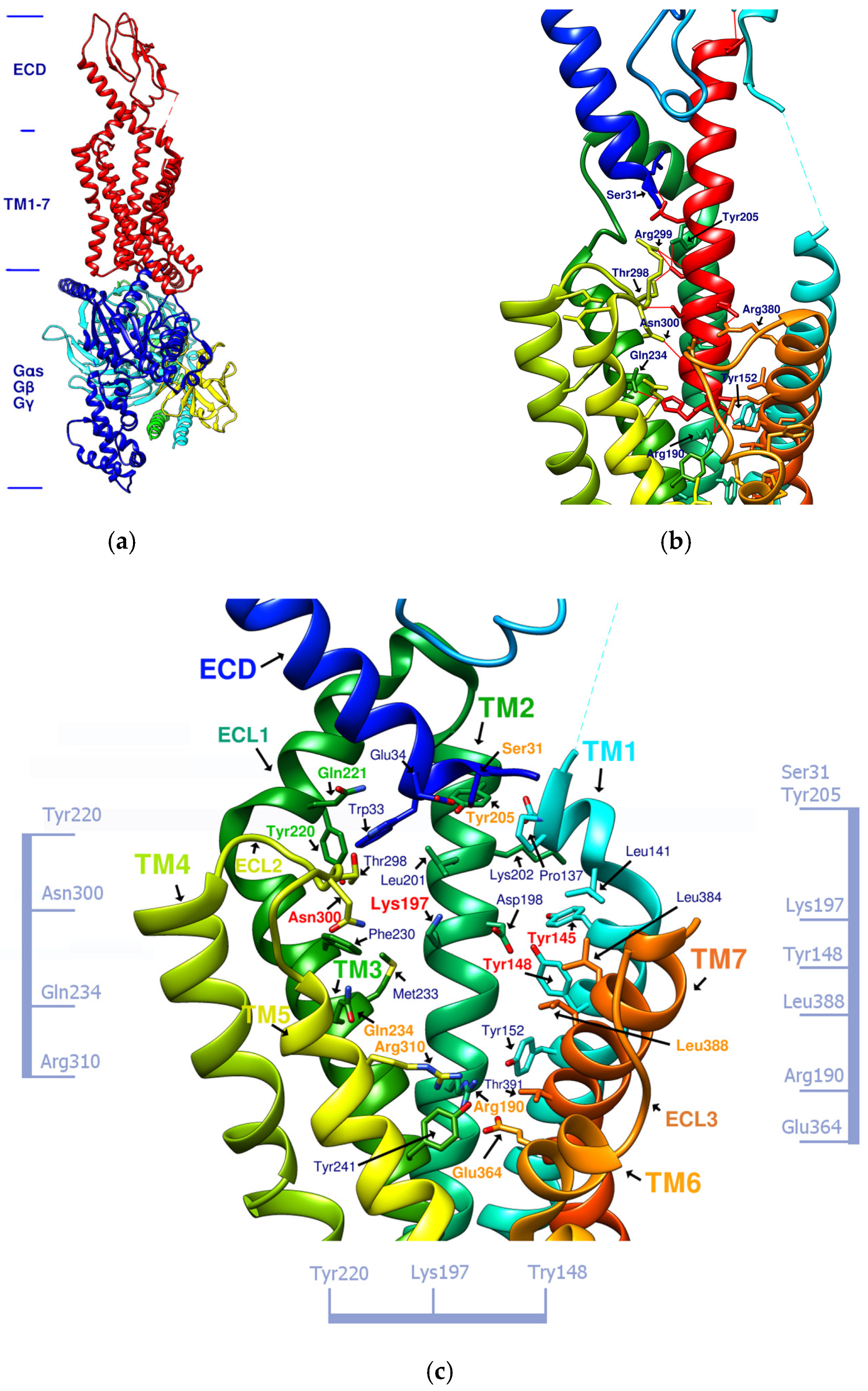
Figure 1. (a) GLP-1R and the G-protein (PDB:6X19). (b) GLP-1 (red alpha helix, the original ligand of GLP-1R)/GLP-1R (transmembrane region) complex (PDB:6X18). (c) Important amino acid residues in the transmembrane region (TM1-7) of GLP-1R (PDB:6X19). Ser31, Tyr205, Gln234, Arg310, Arg190, Glu364, and Leu388, shown in orange, delineate the approximate boundaries of the active site. Asn300, Lys197, Tyr148, and Tyr145, shown in red, are the residues involved in the important hydrogen bonds and π–π interaction network in the middle section. Tyr220 and Gln221, shown in green, are located on ECL1. The labeling of these important amino acids is based on the related studies of GLP-1 agonists [7][18]. The amino acids marked on the vertical axis on both sides and the horizontal axis on the bottom can be used as reference axes for analyzing the configuration of GLP-1R agonists. TM2 can also serve as a reference axis for configurational analysis of GLP-1RAs.
Figure 1b shows that α-helical GLP-1 is inserted vertically from the ECD to TM1-7 in the transmembrane region. From this perspective, all amino acid residues where GLP-1 interacts with GLP-1R can be seen. This perspective best expresses the active site of GLP-1R and is the perspective adopted by most of the relevant literature. Figure 1c lists amino acids on GLP-1R that affect cAMP accumulation or binding affinity. According to studies by K Coopman, D Wootten, and D Yang [19][20][21], essential amino acid residues that affect the binding affinity of GLP-1 or exendin-4 include Y148, Y152, R190, K197, D198, L201, M204, Y205, Q234, Y235, W284, E294, W297, T298, R299, N300, Y305, R310, E364, R380, L384, L388, etc. Residue mutations that may affect cAMP accumulation include Y152, R190, K197, D198, Q234, Y205, W297, N300, R310, E364, T391, etc. These amino acid residues are more numerous than those that interact with GLP-1, and their interactions are also considered when developing new GLP-1R agonists.
2. Development of Small-Molecule Nonpeptide GLP-1R Agonists
The reported GLP-1R agonists include early polypeptide agonists with a secondary structure, truncated free short peptides, and novel nonpeptide small molecules (Figure 2). Although semaglutide has dosage forms for oral administration, the development of small oral nonpeptide GLP-1RA has always been one of the prevalent issues for new drug research and innovation in addition to GLP-1 analogs. According to A Jazayeri et al., it was found that peptide 5 (PDB:5NX2) modified from the GLP-1 truncated sequence (8-17) can produce total agonist activity on GLP-1R [7]. Peptide 5 establishes a hydrogen bond with Ser31, Asn300, Tyr152, Arg190, etc., on GLP-1R, which is similar to the partial interaction between GLP-1 and GLP-1R. The crystal of peptide 5 appears to be a linear peptide and no longer has an α-helical structure. Its top end is fixed with a cap that passes through Trp33 and turns to ECL1 instead of extending to ECD as GLP-1 does (Figure 3) [7]. The GLP-1R–GLP-1–LSN3160440 complex (PDB:6VCB) reveals a novel drug design concept for the activation of GLP-1R [22]. LSN3160440 is an allosteric modulator with molecular glue or uncompetitive pharmacology, which only occupies the local interval between TM1 and TM2 and needs to cooperate with GLP-1 (9–36) NH2 to produce enough effect. At a concentration of 1 μM, it can convert GLP-1 (9–36) NH2 from a 3% effective partial agonist to a full agonist [22]. Recently, nonpeptide GLP-1R agonist PF-06882961, also known as UK4 (PDB:6X1A); LY3502970, also known as OWL-833, V6G (PDB:6XOX and PDB:7E14); and TT-OAD2 (PDB:6ORV) were found entirely out of a GLP-1 peptidomimetic basis. UK1 (PDB:6X19) and V6G (PDB:6XOX) are the ligands of GLP-1R with similar structures [17][18][23][24]. The molecular weights of LY3502970, TT-OAD2, and PF-06882961 are 883.0, 929.7, and 555.6 g/mol, respectively. TT-OAD2 and PF-06882961 have etheric bonds in their structures, which increase the flexibility of the molecule. LY3502970 and UK1 have multiple side chains on the backbone. There are multiple hydrogen bond donors and acceptors in their structures, which are beneficial for interacting with GLP-1R. These molecules bind to the middle-upper regions of the GLP-1R transmembrane helix rather than extending straight to the lower area around Arg190 (Figure 3).
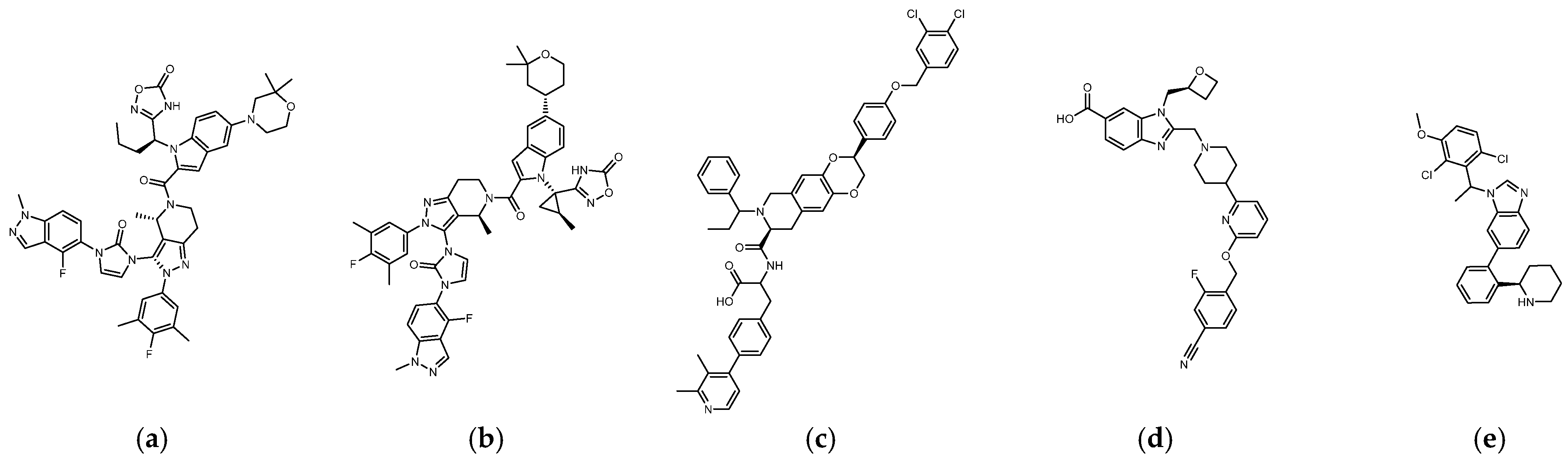
Figure 2. (a) UK1 (PDB:6X19; MW: 886 g/mol), (b) LY3502970 or V6G, OWL-833 (PDB:6XOX; MW: 883.0 g/mol), (c) TT-OAD2 (PDB:6ORV; MW: 929.7 g/mol), (d) PF-06882961 or UK4 (PDB:6X1A; MW: 555.6 g/mol), (e) LSN3160440 or QW7 (PDB:6VCB; MW: 480.43 g/mol).
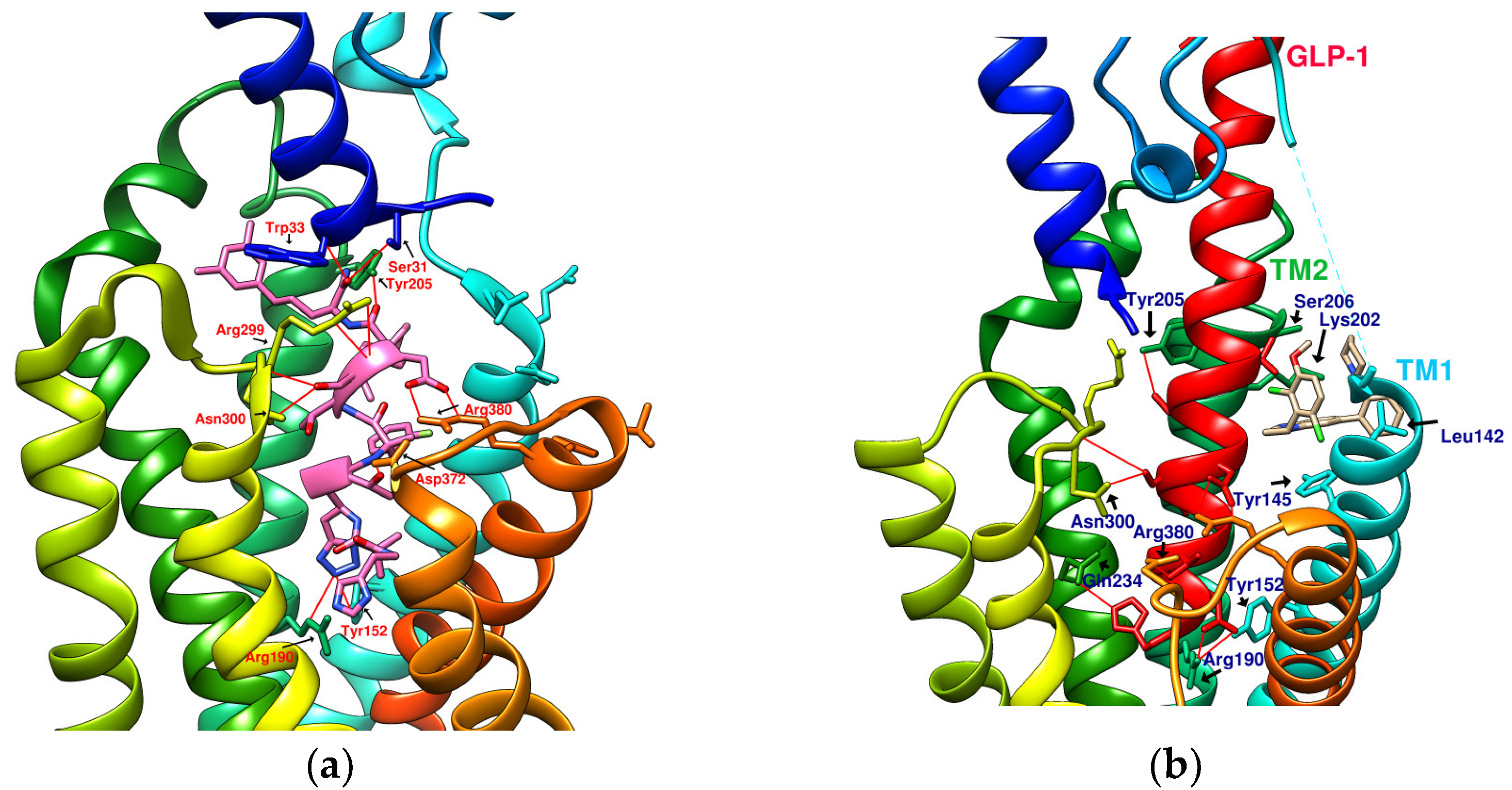
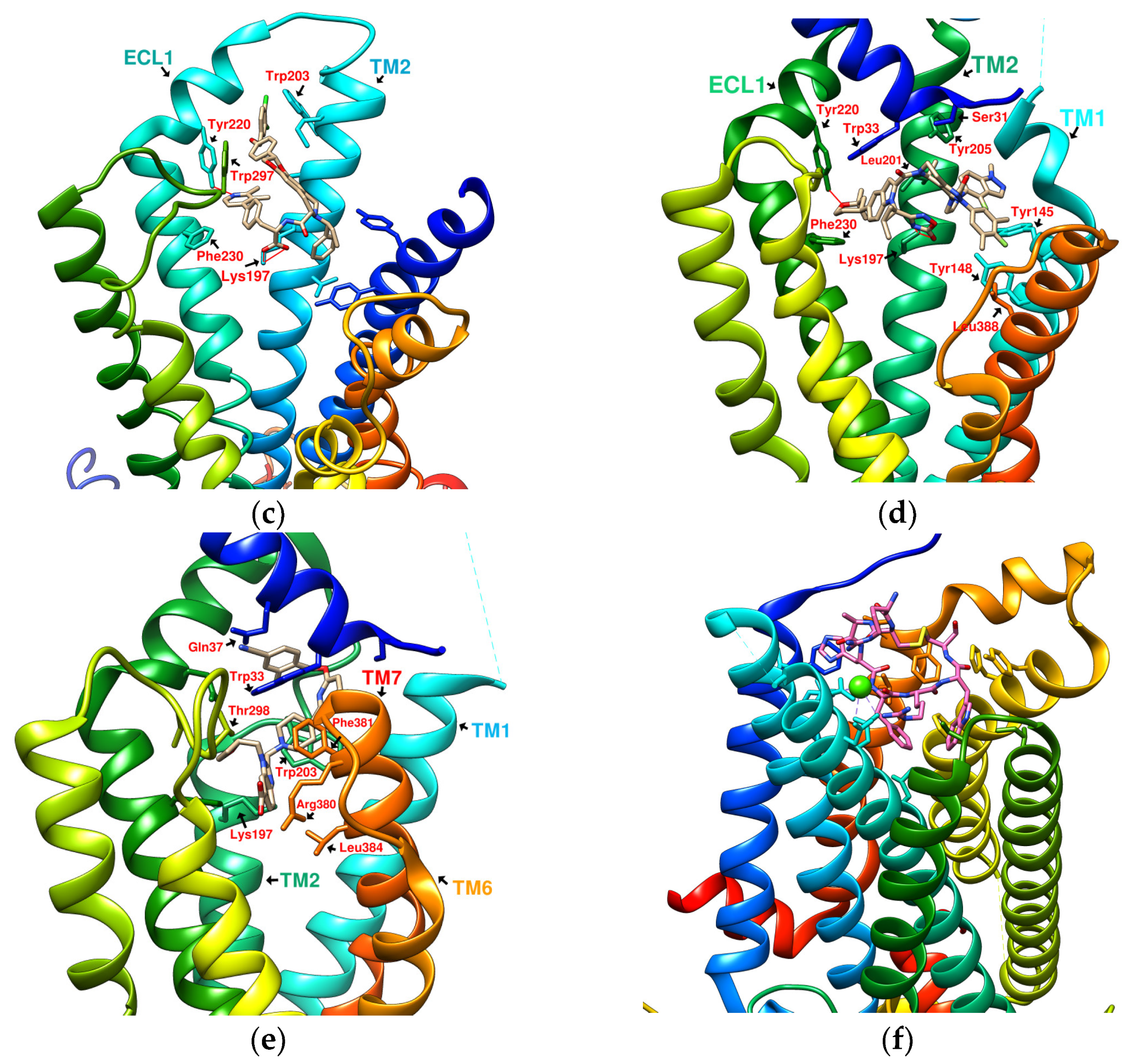
Figure 3. Reference GLP-1R and MC4 (GPCR) complex (PDB file from RCSB Protein Data Bank). (a) Peptide 5/GLP-1R (PDB:5NX2); (b) LSN3160440 and GLP-1 in GLP-1R (PDB:6VCB); (c) TT-OAD2/GLP-1R (PDB:6ORV); (d) LY3502970 in GLP-1R (PDB:6XOX); (e) PF-06882961/GLP-1R (PDB:6X1A); (f) Setmelanotide/MC4R (PDB:7PIU).
TT-OAD2 adopts a U-shaped orientation on GLP-1R. Its backbone extends from Lys197 to Tyr220, sitting between TM2, TM3, and ECL1 [24]. LY3502970 (OWL-833/V6G) [18] displays a three-branch conformation to interact with residues on ECD, TM1, TM2, TM3, and ECL2 (similar to the role of UK1 in PDB:6X19); thus, the range of its influence is greater than that of TT-OAD2. According to T Kawai et al., LY3502970 reacts as a more potent cAMP agonist than TT-OAD2 [18]. The configuration of PF-06882961 in GLP-1R (PDB:6X1A) shows that one end occupies the vicinity of TM7, and the other end reaches ECL1 after passing Try33 of the ECD. LY3502970 shows more binding affinity with GLP-1R than PF-06882961. However, in the study, the concentration-response curves of these two agonists on cAMP accumulation were similar (Figure 3) [17]. These small-molecule agonists may impact Lys197, Tyr145, and Tyr148 in the central area of GLP-1R or establish a connection with the ECL1 region, in which case they need to pass through Trp33 and Thr298. Lys197 is an important residue for these agonists to form hydrogen bonds in the center, and Tyr145, Tyr148, and Trp33 are the primary targets for constructing π–π interactions. These three molecules use different strategies to achieve the activation of GLP-1R, suggesting that the activation of GLP-1R has a certain degree of flexibility. Regarding the space, conformation/configuration, and interaction of these molecules with GLP-1R, there may be more possibilities to design new GLP-1R agonists.
In addition to activating cAMP, PF-06882961 has also been shown to promote β-arrestin recruitment, ERK1/2 phosphorylation, and calcium mobilization. Compared with PF-06882961, these responses in LY3502970 are lacking or extremely weak, and related studies have explained that the difference may be due to more interactions between PF-06882961 and TM7 [17]. β-arrestin plays a role in GPCR desensitization (loss of response caused by prolonged use of agonists). β-Arrestin 1-mediated ERK1/2 activation may also be beneficial in protecting β-cells from apoptosis [3][25]. Since the axes of TM5, TM6, and TM7 on GLP-1R are skewed outwards, small molecules that appear in the upper part of the active site have difficulty interacting with amino acids on these three axes. When one end of the molecule tilts to the middle and down to the vicinity of Leu388 and Thr391, the possibility of interacting with TM5-7 will increase, and the corresponding biased activation may be manifested accordingly, as shown by PF-06882961 (Figure 3e).
3. Reference Case of Cyclic Peptides (Cyclopeptides) as GPCR Agonists/Antagonists
Joakim E. Swedberg et al. found that monocyclic or bicyclic α-conotoxin peptidomimetic chimeras act as potent agonists of GLP-1R [26]. They found effective interactions between cyclopeptides and GLP-1R. However, there is currently no protein crystal structure for this study, and there is no way to see the actual configuration of these cyclopeptides on GLP-1R. Melanocortin 4 receptor (MC4R) provides another case study for exploring GPCR agonists/antagonists. MC4R belongs to class A GPCRs, is located in the hypothalamus area, and plays a vital role in appetite and energy control. The defect of MC4R has been observed to cause a lack of satiety and early onset of severe obesity syndrome. Setmelanotide (Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2, MW: 1,1117.3 g/mol) is a highly potent MC4R agonist. The endogenous ligand for MC4R is the tridecapeptide “α-melanocyte-stimulating hormone”, or α-MSH for short. The structure of setmelanotide includes the α-MSH tetrapeptide pharmacophore (His6-Phe7-Arg8-Trp9) and four other amino acids to form an octacyclic peptide containing a pair of disulfide bonds. Compared with α-MSH, setmelanotide can effectively activate the defective MC4R, which has been proven to help with weight control of the MC4R-variant carrier [27]. SHU9119 is an MC4R antagonist. Its molecular formula “Ac-Nle(4)-c(Asp(5)-2′-Nal(7)-Lys(10))α-MSH(4-10)-NH2” also contains truncated α-MSH. PDB:6W25 and PDB:7PIU show the structures of MC4R in complex with SHU9119 and setmelanotide (Ca2+ as a cofactor), respectively, demonstrating the ability of cyclic peptides (cyclopeptides) to interact well with multiple α-helices of GPCRs (Figure 3f) [28][29]. On the other hand, peptide 5 (PDB:5NX2) consists of a truncated sequence (8–17) from GLP-1 that makes it a GLP-1R agonist, but did not undergo a cyclization process [7].
4. Correlation between GLP-1R Active Site and Accommodated Molecular Size
The linear, multi-branched, or cyclic configurations displayed by various GPCR agonists illustrate that an agonist needs to interact with multiple amino acid residues to activate a GPCR. Therefore, GLP-1RA generally has a specific molecular size. PF-06882961 with an MW of 555.6 g/mol exhibits the characteristics of a complete GLP-1R agonist, whereas LSN3160440 with an MW of 480.43 g/mol is set as an ectopic modulator. GLP-1 agonists with a molecular weight of less than 480 g/mol may not be easy to design. The region’s union, occupied by various GLP-1RA, can be approximately framed by the following amino acids: Ser31, Tyr220, Asn300, Gln234, Arg190, Glu364, Leu388, Pro137, and Leu141. These amino acids define a hemispheric-like region. The area is large enough to accommodate molecules about the size of a decapeptide, such as peptide 5 (PDB:5NX2), or a cyclic peptide of a specific size. If the circumference of the cyclic peptide is within this range, it may have a chance to enter.
5. The Research Potential of Pseudostellaria heterophylla, Linum usitatissimum, and Drymaria diandra as Natural Hypoglycemic Products and GLP-1R Agonists
Several studies have shown that Pseudostellaria heterophylla (Taizishen or P. heterophylla), Linum usitatissimum (L. usitatissimum, flaxseed, flax, linseed), and Drymaria diandra (D. diandra, Drymaria cordata, D. cordata) are three plant products that have the effect of lowering blood sugar [30][31][32][33][34][35]. The three plants have in common that they are rich in cyclopeptides [30][36][37][38][39][40][41]. Their cyclopeptides are called “Caryophyllaceae-type cyclopeptides” (CTCs) in the study by NH Tan et al. [40] or “orbitides” in the survey by PG Arnison et al. [41]. These plant N-C cyclic peptides are mainly monocyclic peptides consisting of hydrophobic amino acids and do not contain disulfide bonds. The main components of P. heterophylla include saponins, polysaccharides, and cyclopeptides. Early herbal pharmacopeias state that P. heterophylla can be used to improve fatigue, polydipsia, lung diseases, and physical fitness [30][36]. In modern times, it appears in Chinese medicine as compound prescriptions for the treatment of type 2 diabetes or as a nutritional supplement for the weak after an illness. The primary ingredients of L. usitatissimum (flaxseed) include alpha-linoleic acid, lignans, and cyclopeptides. Studies have shown that it has antidiabetic, anticancer, antimalaria, cardioprotective, and immunomodulatory properties [37]. Drymaria diandra was used to treat diabetes in the Sikkim area of India. In an experiment conducted by S. Patra et al., Drymaria cordata (D. diandra) methanol extract (DCME) was administered to diabetic rats. It was observed that FBG, HbA1c, serum lipids, and related liver and kidney antioxidant parameters decreased. Compared with the diabetes group, the β-cell density of the pancreatic tissue in the treatment group increased [35].
Targets of hypoglycemia in which natural products are often discussed include PTP1B, α-glucosidase, α-amylase, PPARγ, GLUT4, DPP4, etc. [42]. The volume of cyclopeptides in these three plants is larger than that of flavonoids, polyphenols, and terpenes. The active sites of the DPP4 and GLP-1 receptors, both targets of incretin-based therapy, are large enough to accommodate these plant-derived cyclopeptides [43][44]. The active sites of PTP1B, α-glucosidase, α-amylase, PPARγ, and GLUT4 are small compared to DPP4, and large cyclopeptides are not easily accessible. DPP4 inhibition is a mechanism by which many natural products are involved in promoting hypoglycemic effects. Researchers have previously observed the possible impact of P. heterophylla, flaxseed, and D. diandra cyclopeptides on DPP4 through molecular modeling. Most of the cyclopeptides in these plants could have binding affinities better than −9.0 kcal/mol to DPP4. The docking configuration demonstrates that these cyclopeptides can approach and interact with important amino acids near the catalytic center [45]. Compared to DPP4, the role of natural products on GLP-1R is much less discussed [42]. It may be because general small-molecule natural substances do not easily establish hydrogen bonds with multiple α-helices at the active sites of GPCRs. Cyclopeptides may not be able to configure properly in a small active site, but in the case of DPP4, GLP-1R, etc., with a large active region, cyclopeptides may be able to exert their structural properties [44]. The cyclopeptides are not easy to separate and purify, and GLP-1R does not have commercially available reagents such as DPP4, which increases the threshold for studying the effect of cyclopeptides on GLP-1R. The advent of protein crystals of small, nonpeptidic GLP-1R agonists has provided insight into how small molecules without α-helical structures act at the GLP-1 receptor. Multiple cyclopeptides from these three plants are similar in molecular size to LY3502970 and TT-OAD2. The centroids provided by the ligands of protein crystals make it possible to analyze the interaction of natural compounds with GLP-1R by molecular modeling. In docking, the location of these cyclopeptides at GLP-1R can be observed and compared to the positions of LY3502970 and TT-OAD2 in protein crystallization.
DPP4′s active site is located outside the cell membrane, and molecules do not need to enter the cell membrane to interact with it [44]. According to research, GLP-1R agonists (GLP-1RAs) have a greater impact on hemoglobin A1c (HbA1c), fasting plasma glucose (FPG), blood lipids, and body weight in patients with type 2 diabetes than DPP4 inhibitors [6]. GLP-1R is the target of incretin-based therapy that merits particular emphasis and study. However, the interaction with GLP-1R involves crossing the cell membrane, which is a more complicated process than the interaction with DPP4. Nonpeptide GLP-1R agonists currently in development interact with amino acid residues in the α-helical transmembrane domain of GLP-1R; therefore, the cell membrane permeability of the compound should be considered when designing new GLP-1RAs. These plant-derived cyclopeptides are nonpolar, mainly composed of hydrophobic amino acids, and have no additional glycosylation or methylation modifications. Commonly, nonpolar cyclopeptides enter cells via passive transport [46][47]. Methylated cyclopeptides may exhibit accelerated passive transport when moving across membranes [48]. Unlike passive transport, cell-penetrating peptides (CPPs) consisting of three to four arginine residues and one to two hydrophobic residues can induce deformation of the cell membrane and then be taken up by cells via endocytosis [49]. Plant cyclopeptides that do not have the conditions to constitute a CPP are unlikely to disrupt cell membranes through endocytosis. Unmethylated nonpolar cyclopeptides may stay in the cell membrane for some time before passing through, and this membrane crossing process may provide an opportunity to facilitate their interaction with GLP-1R [50].
This entry is adapted from the peer-reviewed paper 10.3390/metabo12060549
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas. Diabetes Res. Clin. Pract. 2019, 157, 107843.
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157.
- Müller, T.D.; Finan, B.; Bloom, S.; D’Alessio, D.; Drucker, D.J.; Flatt, P.; Fritsche, A.; Gribble, F.; Grill, H.; Habener, J. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130.
- Godinho, R.; Mega, C.; Teixeira-de-Lemos, E.; Carvalho, E.; Teixeira, F.; Fernandes, R.; Reis, F. The Place of Dipeptidyl Peptidase-4 Inhibitors in Type 2 Diabetes Therapeutics: A “Me Too” or “the Special One” Antidiabetic Class? J. Diabetes Res. 2015, 2015, 806979.
- Davies, M.J.; Bianchi, C.; Del Prato, S. Use of incretin-based medications: What do current international recommendations suggest with respect to GLP-1 receptor agonists and DPP-4 inhibitors? Metabolism 2020, 107, 154242.
- Aroda, V.R.; Henry, R.R.; Han, J.; Huang, W.; DeYoung, M.B.; Darsow, T.; Hoogwerf, B.J. Efficacy of GLP-1 receptor agonists and DPP-4 inhibitors: Meta-analysis and systematic review. Clin. Ther. 2012, 34, 1247–1258.E22.
- Jazayeri, A.; Rappas, M.; Brown, A.J.H.; Kean, J.; Errey, J.C.; Robertson, N.J.; Fiez-Vandal, C.; Andrews, S.P.; Congreve, M.; Bortolato, A.; et al. Crystal structure of the GLP-1 receptor bound to a peptide agonist. Nature 2017, 546, 254–258.
- Sharma, D.; Verma, S.; Vaidya, S.; Kalia, K.; Tiwari, V. Recent updates on GLP-1 agonists: Current advancements & challenges. Biomed. Pharm. 2018, 108, 952–962.
- Dailey, M.J.; Moran, T.H. Glucagon-like peptide 1 and appetite. Trends Endocrinol. Metab. 2013, 24, 85–91.
- Nuffer, W.A.; Trujillo, J.M. Liraglutide: A New Option for the Treatment of Obesity. Pharmacotherapy 2015, 35, 926–934.
- O’Neil, P.M.; Birkenfeld, A.L.; McGowan, B.; Mosenzon, O.; Pedersen, S.D.; Wharton, S.; Carson, C.G.; Jepsen, C.H.; Kabisch, M.; Wilding, J.P.H. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: A randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet 2018, 392, 637–649.
- Koska, J.; Sands, M.; Burciu, C.; D’Souza, K.M.; Raravikar, K.; Liu, J.; Truran, S.; Franco, D.A.; Schwartz, E.A.; Schwenke, D.C.; et al. Exenatide Protects Against Glucose- and Lipid-Induced Endothelial Dysfunction: Evidence for Direct Vasodilation Effect of GLP-1 Receptor Agonists in Humans. Diabetes 2015, 64, 2624–2635.
- Sloan, L.A. Review of glucagon-like peptide-1 receptor agonists for the treatment of type 2 diabetes mellitus in patients with chronic kidney disease and their renal effects. J. Diabetes 2019, 11, 938–948.
- DeFronzo, R.A.; Ratner, R.E.; Han, J.; Kim, D.D.; Fineman, M.S.; Baron, A.D. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005, 28, 1092–1100.
- Knudsen, L.B.; Lau, J. The Discovery and Development of Liraglutide and Semaglutide. Front. Endocrinol. 2019, 10, 155.
- Manandhar, B.; Ahn, J.M. Glucagon-like peptide-1 (GLP-1) analogs: Recent advances, new possibilities, and therapeutic implications. J. Med. Chem. 2015, 58, 1020–1037.
- Zhang, X.; Belousoff, M.J.; Zhao, P.; Kooistra, A.J.; Truong, T.T.; Ang, S.Y.; Underwood, C.R.; Egebjerg, T.; Šenel, P.; Stewart, G.D.; et al. Differential GLP-1R Binding and Activation by Peptide and Non-peptide Agonists. Mol. Cell 2020, 80, 485–500.e487.
- Kawai, T.; Sun, B.; Yoshino, H.; Feng, D.; Suzuki, Y.; Fukazawa, M.; Nagao, S.; Wainscott, D.B.; Showalter, A.D.; Droz, B.A.; et al. Structural basis for GLP-1 receptor activation by LY3502970, an orally active nonpeptide agonist. Proc. Natl. Acad. Sci. USA 2020, 117, 29959–29967.
- Coopman, K.; Wallis, R.; Robb, G.; Brown, A.J.; Wilkinson, G.F.; Timms, D.; Willars, G.B. Residues within the transmembrane domain of the glucagon-like peptide-1 receptor involved in ligand binding and receptor activation: Modelling the ligand-bound receptor. Mol. Endocrinol. 2011, 25, 1804–1818.
- Wootten, D.; Reynolds, C.A.; Smith, K.J.; Mobarec, J.C.; Koole, C.; Savage, E.E.; Pabreja, K.; Simms, J.; Sridhar, R.; Furness, S.G.B.; et al. The Extracellular Surface of the GLP-1 Receptor Is a Molecular Trigger for Biased Agonism. Cell 2016, 165, 1632–1643.
- Yang, D.; de Graaf, C.; Yang, L.; Song, G.; Dai, A.; Cai, X.; Feng, Y.; Reedtz-Runge, S.; Hanson, M.A.; Yang, H.; et al. Structural Determinants of Binding the Seven-transmembrane Domain of the Glucagon-like Peptide-1 Receptor (GLP-1R). J. Biol. Chem. 2016, 291, 12991–13004.
- Bueno, A.B.; Sun, B.; Willard, F.S.; Feng, D.; Ho, J.D.; Wainscott, D.B.; Showalter, A.D.; Vieth, M.; Chen, Q.; Stutsman, C.; et al. Structural insights into probe-dependent positive allosterism of the GLP-1 receptor. Nat. Chem. Biol. 2020, 16, 1105–1110.
- Cong, Z.; Chen, L.N.; Ma, H.; Zhou, Q.; Zou, X.; Ye, C.; Dai, A.; Liu, Q.; Huang, W.; Sun, X.; et al. Molecular insights into ago-allosteric modulation of the human glucagon-like peptide-1 receptor. Nat. Commun. 2021, 12, 3763.
- Zhao, P.; Liang, Y.L.; Belousoff, M.J.; Deganutti, G.; Fletcher, M.M.; Willard, F.S.; Bell, M.G.; Christe, M.E.; Sloop, K.W.; Inoue, A.; et al. Activation of the GLP-1 receptor by a non-peptidic agonist. Nature 2020, 577, 432–436.
- Quoyer, J.; Longuet, C.; Broca, C.; Linck, N.; Costes, S.; Varin, E.; Bockaert, J.; Bertrand, G.; Dalle, S. GLP-1 mediates antiapoptotic effect by phosphorylating Bad through a beta-arrestin 1-mediated ERK1/2 activation in pancreatic beta-cells. J. Biol. Chem. 2010, 285, 1989–2002.
- Swedberg, J.E.; Schroeder, C.I.; Mitchell, J.M.; Durek, T.; Fairlie, D.P.; Edmonds, D.J.; Griffith, D.A.; Ruggeri, R.B.; Derksen, D.R.; Loria, P.M. Cyclic alpha-conotoxin peptidomimetic chimeras as potent GLP-1R agonists. Eur. J. Med. Chem. 2015, 103, 175–184.
- Collet, T.H.; Dubern, B.; Mokrosinski, J.; Connors, H.; Keogh, J.M.; Mendes de Oliveira, E.; Henning, E.; Poitou-Bernert, C.; Oppert, J.M.; Tounian, P.; et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol. Metab. 2017, 6, 1321–1329.
- Yu, J.; Gimenez, L.E.; Hernandez, C.C.; Wu, Y.; Wein, A.H.; Han, G.W.; McClary, K.; Mittal, S.R.; Burdsall, K.; Stauch, B.; et al. Determination of the melanocortin-4 receptor structure identifies Ca(2+) as a cofactor for ligand binding. Science 2020, 368, 428–433.
- Heyder, N.A.; Kleinau, G.; Speck, D.; Schmidt, A.; Paisdzior, S.; Szczepek, M.; Bauer, B.; Koch, A.; Gallandi, M.; Kwiatkowski, D.; et al. Structures of active melanocortin-4 receptor-Gs-protein complexes with NDP-α-MSH and setmelanotide. Cell Res. 2021, 31, 1176–1189.
- Hu, D.J.; Shakerian, F.; Zhao, J.; Li, S.P. Chemistry, pharmacology and analysis of Pseudostellaria heterophylla: A mini-review. Chin. Med. 2019, 14, 21.
- Su, K.; Zhu, F.; Guo, L.; Zhu, Y.; Li, W.; Xiong, X. Retrospective study on Professor Zhongying Zhou’s experience in Traditional Chinese Medicine treatment on diabetic nephropathy. J. Tradit. Chin. Med. 2013, 33, 262–267.
- Chen, J.; Pang, W.; Shi, W.; Yang, B.; Kan, Y.; He, Z.; Hu, J. Structural Elucidation of a Novel Polysaccharide from Pseudostellaria heterophylla and Stimulating Glucose Uptake in Cells and Distributing in Rats by Oral. Molecules 2016, 21, 1233.
- Mani, U.V.; Mani, I.; Biswas, M.; Kumar, S.N. An open-label study on the effect of flax seed powder (Linum usitatissimum) supplementation in the management of diabetes mellitus. J. Diet. Suppl. 2011, 8, 257–265.
- Pan, A.; Sun, J.; Chen, Y.; Ye, X.; Li, H.; Yu, Z.; Wang, Y.; Gu, W.; Zhang, X.; Chen, X.; et al. Effects of a flaxseed-derived lignan supplement in type 2 diabetic patients: A randomized, double-blind, cross-over trial. PLoS ONE 2007, 2, e1148.
- Patra, S.; Bhattacharya, S.; Bala, A.; Haldar, P.K. Antidiabetic effect of Drymaria cordata leaf against streptozotocin-nicotinamide-induced diabetic albino rats. J. Adv. Pharm. Technol. Res. 2020, 11, 44–52.
- Lu, F.; Yang, H.; Lin, S.D.; Zhao, L.; Jiang, C.; Chen, Z.B.; Liu, Y.Y.; Kan, Y.J.; Hu, J.; Pang, W.S. Cyclic Peptide Extracts Derived From Pseudostellaria heterophylla Ameliorates COPD via Regulation of the TLR4/MyD88 Pathway Proteins. Front. Pharmacol. 2020, 11, 850.
- Shim, Y.Y.; Song, Z.; Jadhav, P.D.; Reaney, M.J. Orbitides from flaxseed (Linum usitatissimum L.): A comprehensive review. Trends Food Sci. Technol. 2019, 93, 197–211.
- Hsieh, P.W.; Chang, F.R.; Wu, C.C.; Wu, K.Y.; Li, C.M.; Wang, W.Y.; Gu, L.C.; Wu, Y.C. Selective Inhibition of Collagen-Induced Platelet Aggregation by a Cyclic Peptide from Drymaria diandra. Helv. Chim. Acta 2004, 87, 57–66.
- Hsieh, P.-W.; Chang, F.-R.; Lee, K.-H.; Hwang, T.-L.; Chang, S.-M.; Wu, Y.-C. A new anti-HIV alkaloid, drymaritin, and a new C-Glycoside flavonoid, diandraflavone, from Drymaria d iandra. J. Nat. Prod. 2004, 67, 1175–1177.
- Tan, N.H.; Zhou, J. Plant cyclopeptides. Chem. Rev. 2006, 106, 840–895.
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160.
- He, J.-H.; Chen, L.-X.; Li, H. Progress in the discovery of naturally occurring anti-diabetic drugs and in the identification of their molecular targets. Fitoterapia 2019, 134, 270–289.
- Nauck, M.A. Incretin-based therapies for type 2 diabetes mellitus: Properties, functions, and clinical implications. Am. J. Med. 2011, 124, S3–S18.
- Klemann, C.; Wagner, L.; Stephan, M.; von Hörsten, S. Cut to the chase: A review of CD26/dipeptidyl peptidase-4’s (DPP4) entanglement in the immune system. Clin. Exp. Immunol. 2016, 185, 1–21.
- Liao, H.-J.; Tzen, J.T.C. The Potential Role of Cyclopeptides from Pseudostellaria heterophylla, Linum usitatissimum and Drymaria diandra, and Peptides Derived from Heterophyllin B as Dipeptidyl Peptidase IV Inhibitors for the Treatment of Type 2 Diabetes: An In Silico Study. Metabolites 2022, 12, 387.
- Bockus, A.T.; McEwen, C.M.; Lokey, R.S. Form and function in cyclic peptide natural products: A pharmacokinetic perspective. Curr. Top. Med. Chem. 2013, 13, 821–836.
- Ahlbach, C.L.; Lexa, K.W.; Bockus, A.T.; Chen, V.; Crews, P.; Jacobson, M.P.; Lokey, R.S. Beyond cyclosporine A: Conformation-dependent passive membrane permeabilities of cyclic peptide natural products. Future Med. Chem. 2015, 7, 2121–2130.
- Räder, A.F.B.; Reichart, F.; Weinmüller, M.; Kessler, H. Improving oral bioavailability of cyclic peptides by N-methylation. Bioorg Med. Chem. 2018, 26, 2766–2773.
- Dougherty, P.G.; Sahni, A.; Pei, D. Understanding Cell Penetration of Cyclic Peptides. Chem. Rev. 2019, 119, 10241–10287.
- Wang, C.K.; Craik, D.J. Cyclic peptide oral bioavailability: Lessons from the past. Biopolymers 2016, 106, 901–909.
This entry is offline, you can click here to edit this entry!