Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
DDK is formed by the association of the kinase subunit, cell division cycle 7 (Cdc7), with its activating partner, dumb-bell factor 4 (Dbf4). By analogy with CDK, Cdc7:Dbf4 is now most often referred to as ‘Dbf4-dependent kinase’ or DDK.
- DDK
- CDK
- DNA replication
1. Introduction: Ensuring Chromosome Maintenance during the Mitotic Cell Division Cycle
In addition to the faithful duplication of the genome that occurs during S phase, the pre-replicative chromatin environment must also be re-established to maintain cell identity. Furthermore, in order to ensure the equal segregation of the duplicated genome to progeny cells, the replicated chromatid pairs must remain tethered prior to their anaphase segregation. The successful inheritance of a chromosome during cell division therefore requires the coordination of DNA replication and repair, chromatin re-formation and the establishment of chromatid cohesion. These activities should occur in concert to allow chromosome segregation and cell division during mitosis, all within a background of ongoing cellular processes, such as gene transcription, protein translation and energy metabolism.
The two kinases, cyclin-dependent kinase, CDK, and the Dbf4-dependent kinase, DDK, are required for DNA replication initiation (Figure 1 and Figure 2) [1,2,3,4]. CDK functions as the ‘global’ regulator of S phase entry and progression whilst ‘local’ control of replication origin activation is delegated to DDK [5]. This delegation of responsibility to DDK by CDK in higher eukaryotes is mediated by the CDK-dependent transcription of DDK and therefore its activation, at the G1-to-S transition [6,7,8]. In addition to its well characterised role in the initiation of DNA replication, it has been shown that DDK is required for chromatin re-formation following replication fork progression [9,10,11], for replication fork stability and DNA repair [12,13,14,15], and for the establishment of replicated chromatid cohesion [16,17] (Figure 1). The maintenance of chromosome stability during the mitotic cell-cycle therefore requires the coordination of CDK and DDK activity. This delegation of responsibility, or outsourcing, to DDK by CDK allows CDK to maintain outright control of S phase progression and the cell-cycle phase transitions whilst permitting ongoing chromatin replication and cohesion establishment to be completed and achieved faithfully.
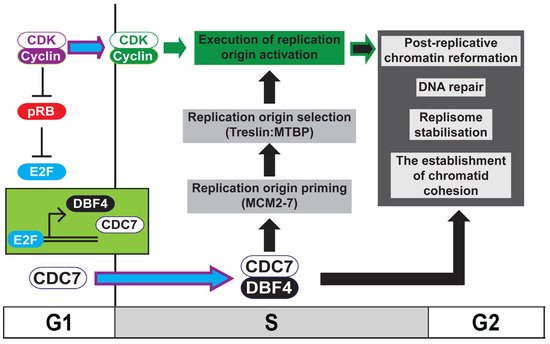
Figure 1. Model for the role of DDK in ensuring chromosome stability. Activation of Cdc7 and Dbf4 transcription by E2F in higher eukaryotes occurs following the inactivation of pRB by rising CDK levels at the G1-to-S transition. During S phase, the activation of replication origins requires the coordination between active DDK (Cdc7:Dbf4), which both primes (MCM2-7 phosphorylation) and directs origin selection (Treslin:MTBP phosphorylation) and CDK, which executes replication initiation. In addition to its well-characterised role in replication initiation, DDK is required for the establishment of replicated chromatid cohesion, the maintenance of replisome stability and DNA repair, and is required for the re-formation of chromatin following replication of the DNA. The coordination between CDK and DDK therefore ensures chromosome maintenance during the mitotic cell-cycle.
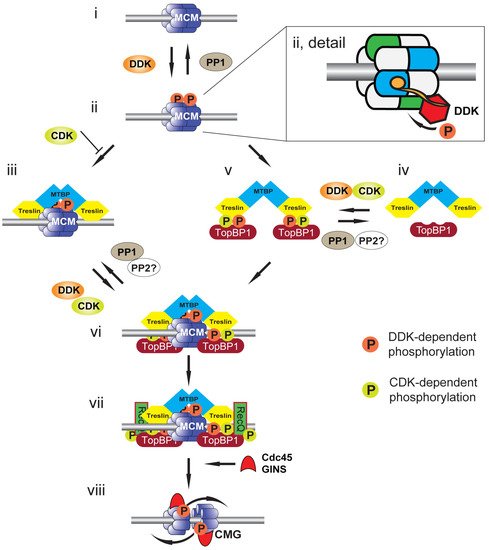
Figure 2. Model for CMG formation and activation. Cartoon illustrating events at a replication origin leading to replication initiation. (i) Replication origin DNA (grey bar) licensed by DNA-bound MCM2-7 double hexamer (blue). (ii) DNA-bound MCM2-7 is phosphorylated by DDK (orange circles) and phosphorylation is reversed by PP1 recruited to origins by Rif1. Detail shows a DNA-bound MCM2-7 double hexamer in which the BRCT phosphobinding-domain of Dbf4 (orange circle) is bound to MCM2 (blue) of one hexamer to localise Cdc7 (red) to phosphorylate MCM4 (green) of the second hexamer. (iii) Treslin:MTBP is recruited to DDK-phosphorylated MCM2-7. This interaction is opposed by CDK (green circles), most likely due to CDK phosphorylation of Treslin. (iv) Soluble Treslin:MTBP can be phosphorylated by both CDK and DDK. (v) Phosphorylated Treslin:MTBP binds to TopBP1 in solution. (vi) The Treslin:MTBP:TopBP1 complex is recruited to DDK-phosphorylated MCM2-7. (vii) RecQ4, the second CDK substrate required for replication initiation, is recruited to MCM2-7:Treslin:MTBP:TopBP1 to form the pre-initiation complex (pre-IC). (viii) The CMG helicase is formed upon Cdc45 and GINS recruitment to MCM2-7. The MCM2-7 double hexamer is split and the helicases overtake one another to support bidirectional DNA replication.
2. Introducing the Dbf4-Dependent Kinase: DDK
DDK is formed by the association of the kinase subunit, cell division cycle 7 (Cdc7), with its activating partner, dumb-bell factor 4 (Dbf4) [18]. By analogy with CDK, Cdc7:Dbf4 is now most often referred to as ‘Dbf4-dependent kinase’ or DDK [19].
Whereas both proteins are well conserved across eukaryotes and orthologs have also been identified in plants, the nomenclature is not as well maintained between organisms (Table 1): with the exception of fission yeast in which the term hsk1 (homolog of seven kinase) is used, ‘Cdc7’ is remarkably well preserved; in contrast, orthologs of Dbf4 are variously known as dfp1 (dumb-bell factor pombe), him1 (hsk-interacting molecule) and rad35 in fission yeast, ASK (activator of S kinase) in human and chiffon in fruit fly. It should be noted, however, that the chiffon locus in fly is unique to that organism and encodes 2 polypeptides, chiffon A (Dbf4-like) and chiffon B (a Gcn5 acetylase binding protein) [20].
Table 1. Matched nomenclature for protein names in human and Xenopus laevis with those of budding yeast, Saccharomyces cerevisiae, fission yeast, Schizosaccharomyces pombe and fruit fly, Drosophila melanogaster.
| Human & Xenopus laevis | S. cerevisiae, S. pombe & D melanogaster |
|---|---|
| Cdc7 | Cdc7 (Sc), Hsk1 (Sp), Cdc7 (Dm) |
| Dbf4, ASK (Hs) | Dbf4 (Sc), dfp1, him1, rad35 (Sp), Chiffon A (Dm) |
| Drf1, Dbf4B (Hs) | - |
| Treslin | Sld3 (Sc) |
| MTBP | Sld7 (Sc) |
| RecQ4 | Sld2 (Sc) |
| Claspin | Mrc1 (Sc) |
| Timeless | Tof1 (Sc) |
| Tipin | Csm3 (Sc) |
| Chk1 | Rad53 (Sc) |
A second Dbf4 paralog, named either Drf1 (Dbf4-related factor 1) or Dbf4B, present in vertebrates including Xenopus, mouse and chicken, was first identified in human cells [21,22,23]. Immediately post-fertilisation, in developing Xenopus laevis eggs, DDK activity is predominantly supported by Drf1, although Dbf4 is also present but in substantially lower abundance [24]. On completion of the 12 reductive cleavage divisions immediately following fertilisation, eggs pass through the mid-blastula transition (MBT). At the MBT, gross reorganization of the cell-cycle occurs: bulk transcription begins, S phase and therefore cell-cycle length are extended and cell-cycle checkpoints are established [25,26,27]. Activation of Chk1 following the MBT promotes SCFβTRCP degradation of Drf1, enabling the switch to Dbf4, which mediates the change in S phase kinetics [28].
In order to maximise comprehensibility in this review, we will, where possible, use the most commonly used names: Cdc7, Dbf4 and Drf1.
3. Cell-Cycle Regulation of DDK Activity
Early studies in budding yeast by Hartwell and colleagues of the execution point of the various cell division cycle mutants showed Cdc7 functioning downstream of the CDK kinase subunit Cdc28 and downstream of the ubiquitylation substrate adapter protein Cdc4; it was also reported that protein synthesis was required after these arrest points for the initiation of DNA replication to occur [29,30]. Akin to the cyclin:CDK analogy, it is the cell-cycle regulated abundance of Dbf4 that restricts the activation of DDK to the G1-to-S transition, with Cdc7 being present throughout the cell-cycle in this organism [31]. The periodicity of DDK activity is regulated not only by Dbf4 gene transcription, but also by cell-cycle-dependent protein degradation [32,33,34,35].
In contrast, in the human cell lines studied, it has been shown that the abundance of both Cdc7 and Dbf4/Drf1 protein levels are periodic [36,37]. The peak of Dbf4 (and also Drf1) mRNA is in the late G1 and S phase [21,38,39]. In mammalian cells, both Cdc7 and Dbf4/Drf1 are transcriptional targets of the E2F family of transcription factors, activated following satisfaction of the restriction point by either pRb inactivation, degradation or both, upon activation of CDK during G1 [6,7,8]. Consistent with this, Cdc7 and Dbf4/Drf1 proteins levels rapidly increase as cells enter S phase and high levels are maintained in G2 and early M [21,36,37]. That CDK is required for the transcriptional activation of its partner S phase kinase DDK in human cells illustrates the hierarchical nature of their relationship (Figure 1).
Following passage of the metaphase-to-anaphase transition and through until mid-G1, Cdc7 protein levels in human cells and Dbf4/Drf1 protein levels in both yeast and human return to their initial low levels [32,34,35,36,37]. This stark drop in levels of the kinase activating subunit in budding yeast is mediated by the APC/Cyclosome (APC/C) [33,34,35]. Consistent with this, levels of Dbf4 remain low in α-factor (G1) arrested budding yeast and in quiescent human cells [21,33,34,35,39]. Inactivation of the APC/C in G1 mediated by G1/S CDK and Skp1:Cullin1:F Box:Cdc4 allows the accumulation of DDK activity as budding yeast cells pass into S phase. In human cells, inactivation of APC/C:Cdh1 by Chk1 following replication stress stabilises both Cdc7 and Dbf4 suggesting that DDK is also a target of Cdh1 in higher eukaryotes [40].
4. Replication Origin Priming: DDK-Mediated Phosphorylation of MCM2-7
During late mitosis (telophase) and G1, potential replication origins are first loaded with inactive double hexamers of the MCM2-7 proteins to license them for a single initiation event in the upcoming S phase [1,2,4]. DDK plays a key role in phosphorylating MCM2-7 double hexamers to drive their transformation into the replicative CMG (Cdc45:MCM:GINS) helicase. Initial studies of Cdc7 and Dbf4 function in yeast identified MCM subunits as suppressors of DDK temperature sensitive mutations and localized DDK to replication origins [41,42]. Subsequent studies in human cells provided the first direct link between DDK and MCM2-7 phosphorylation by showing in vitro that 2 MCM subunits were DDK substrates [43]. In budding yeast, human and in Xenopus cell-free extracts, phosphorylation of MCMs 2, 4 and 6 have been shown to be DDK-dependent [12,44,45,46,47,48]. It remained unclear, however, which of these were the key physiological targets of DDK to support DNA replication in vivo.
In budding yeast, in vivo, the N-terminus of MCM4 is hyperphosphorylated by DDK [45]. An autoinhibitory activity present in the N-terminus of MCM4 is relieved upon DDK phosphorylation and in yeast cells lacking this region, DDK is not required for cell viability [45]. This suggests that relief of MCM4 autoinhibition by its phosphorylation is the essential function of DDK in this organism to support DNA replication initiation. The precise role of the phosphorylation of the other MCM subunits remains to be determined but may be important for other functions of DDK in maintaining chromosome stability (see below).
Consistent with the identification of MCM4 as the key target of DDK-mediated phosphorylation in budding yeast, it was found that in both human cells and in the Xenopus cell-free system that hyperphosphorylation of MCM4, but not the phosphorylation of specific MCM2 residues, correlates with replication initiation [12,49]. In these studies, MCM4 hyperphosphorylation occurred only on chromatin-bound double hexameric MCM2-7 at licensed replication origins. In contrast, phosphorylation of two known DDK target residues on MCM2 (S40 and S53) occurred on single hexamers free in the nucleoplasm and could be inhibited at concentrations of DDK inhibitors that do not inhibit DNA replication. This suggests that the MCM2-7 double hexamer at licensed replication origins provides the key substrate for DDK to support replication initiation.
The recent description of structural models of DDK associated with chromatin-bound MCM2-7 has highlighted the key role of double hexamer formation on chromatin in promoting the activating phosphorylation of MCM4 [50,51,52]. Dbf4 docks onto one MCM2-7 hexamer to support the phosphorylation of the other hexamer (Figure 2ii, detail). Loss of the Dbf4 docking domain abrogates phosphorylation. The rotationally symmetrical nature of the hexamer explains the ability to phosphorylate both MCM4 subunits at the origin. The docking of Dbf4 to the double hexamer is mediated by an N-terminal BRCT (BRCA1 C-terminal) phosphobinding motif; this motif is conserved in higher eukaryotic Dbf4 orthologs. The docking site for the Dbf4 BRCT domain on the hexamer is the phosphorylated N-terminus of MCM2, although the phosphorylating kinase remains to be determined. Two candidate kinases are CDK and DDK itself: it may be that the previously described DDK-mediated phosphorylation of S40 and S53 of MCM2 in single MCM2-7 hexamers promotes this association on formation of the double hexamer on chromatin; alternatively, or in addition to this, CDK may mediate this phosphorylation to signal fitness for S phase entry.
The phosphorylation of MCM4 by DDK is required to support the localization of key replication proteins to MCM2-7 [1,2,4] (Figure 2). In budding yeast, DDK phosphorylation of MCM2-7 promotes the recruitment of the Sld3:Sld7 complex and Cdc45 to origins. The combination of DDK activity and CDK, which phosphorylates Sld3 and Sld2, promotes the association of Dpb11 and Sld2 with Sld3:Sld7, facilitating the association of the GINS complex and DNA polymerase ε to origin sites. In combination, these factors recruited to MCM2-7 at licensed origins, together with MCM10, promote the formation and separation of the 2 CMG replicative helicases that then function to support DNA unwinding, replication initiation and elongation. Since loss of the auto-inhibitory N-terminal domain of MCM4 in budding yeast permits replication initiation in the absence of DDK and mutation of MCM5 supresses the requirement for DDK in replication initiation, this suggests that MCM2-7 and possibly just MCM4, are the only essential DDK substrates for the initiation of replication in budding yeast.
The role of DDK-mediated phosphorylation of the chromatin-bound MCM2-7 double hexamer in promoting replication initiation has been shown to be substantially conserved in higher eukaryotes (Figure 2). CDK-dependent phosphorylation of Treslin (TopBP1-interacting, replication-stimulating protein), the metazoan ortholog of Sld3, is required for its association with TopBP1, the metazoan ortholog of Dpb11 [53,54,55]. In the Xenopus cell-free system, the association of chromatin-bound MCM2-7 double hexamers with the Treslin:MTBP (MDM2-binding protein) complex, the ortholog of the budding yeast Sld3:Sld7 complex, is both increased and strengthened following DDK phosphorylation of MCM4 [5].
5. Rif1: Antagonising DDK-Mediated MCM4 Phosphorylation
Whereas DDK mediates MCM4 phosphorylation to support replication initiation, recent studies have identified a role for protein phosphatase 1 (PP1) in the reversal of this [47,56,57,58] (Figure 2i). In human cells and in Xenopus egg extract, PP1 continually dephosphorylates MCM4 such that inhibition of DDK mid-S phase inhibits further replication initiation [12,47,59]. In yeast, human cells and in the Xenopus cell-free system, PP1 is targeted to MCM2-7 on chromatin by Rif1 (Rap1-interacting factor 1) [12,56,59]. Rif1 was first identified as an antagonist of Rap1-mediated gene silencing [60]. PP1 binds Rif1 by the PP1-targeting motif RVxF [61,62]. Mutation of these residues or loss of Rif1 leads to the increased activation of replication origins and compensates for a reduction in DDK activity, facilitating the shortening of S phase, showing that whereas DDK promotes S phase progression, Rif1 counteracts this [12,13,56,59].
Loss of Rif1 or the Rif1:PP1 interaction also perturbs the replication timing programme [12,63,64]. This result is consistent with the idea that selection of origins for DDK-dependent phosphorylation forms part of the mechanism that enacts the replication timing programme. In addition to replication timing, Rif1 has been shown to impact nuclear architecture, limiting interaction between domains with the same replication timing [64]. Both nuclear organization and replication timing depend upon Rif1:PP1 interaction; however, timing can be established and executed independent of a specific 3D genome organization or spatial distribution of replication foci [65]. This suggests that coregulation of replication timing and genome organization is specifically linked to Rif1 and not generic nuclear architecture.
In addition to this, Rif1:PP1 has been found to function in a number of DNA replication-associated processes including telomere length regulation, DNA repair and the promotion of replication licensing in G1 by stabilizing Orc1 [59,60,66].
It should be noted that a recent study describes the growth of both human and murine cells in which DDK activity is either inhibited or depleted [67]. Murine cells harbouring an ‘analogue sensitive’ mutation in the Cdc7 ATP binding pocket showed retarded proliferation but did not arrest following treatment with an ATP analogue; furthermore, following either chemically- or genetically-mediated degradation of either Cdc7 or Dbf4 (a requirement for Drf1 was not tested), both human and murine cells can maintain proliferation although changes to S phase length were apparent. The authors conclude that DDK activity is not strictly necessary for proliferation and that it is functionally redundant with CDK. However, although DDK proteins levels were significantly reduced following induced degradation the targeted proteins remained detectable and furthermore, although the phosphorylation of a specific MCM2 residues was lost upon degradation, the phosphorylation of residues in the N-terminal region of MCM4, which better correlates with replication initiation, was still apparent. Thus, it may be the case that proliferation in these cells is the result of a combination of the remaining DDK activity and the maintained phosphorylation of DDK target sites, perhaps following the inactivation of Rif1:PP1 by CDK [68]. Consistent with this, the loss of Rif1 reduces the sensitivity of cells to DDK inhibition [69]. In order to clarify these issues, further work is needed to determine the essential phosphorylation sites required for cell proliferation and which kinases can phosphorylate them.
This entry is adapted from the peer-reviewed paper 10.3390/biology11060877
This entry is offline, you can click here to edit this entry!