Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Medicinal
Quinazolines are the most versatile, ubiquitous and privileged nitrogen bearing heterocyclic compounds with a wide array of biological and pharmacological applications. Most of the anti-cancer agents featuring quinazoline pharmacophore have shown promising therapeutic activity.
- quinazoline
- histone deacetylases (HDAC)
- cancer
- kinase receptor
- multi drug resistance
1. History and Significance of Quinazolines
Heterocyclic compounds and, in particular, nitrogen bearing compounds have tremendous values in the field of medicinal chemistry. With a plethora of potential pharmacological activities, nitrogen containing derivatives continue to enthrall scientists worldwide with their therapeutic properties, and this is the reason why heterocyclic compounds are the building blocks of many drugs and pharmaceutical products [4,5,6,7]. Among the nitrogen containing heterocyclic aromatic scaffolds, quinazolines are considered as privileged compounds. Quinazoline scaffolds represent the ubiquitous class of heterocyclic compounds that are found in over 200 naturally occurring alkaloids [4,8,9]. They have gained wide recognition and also constitute the most studied moieties in heterocyclic chemistry. Ever since their discovery, these fused ring compounds have been the center of interest for medicinal chemists due to their attractive biological and pharmacological applications. Although quinazolines were first recognized for their anti-malarial activity, their derivatives later found diverse applications such as antimicrobial, anti-fungal, anti-convulsant, anti-tumor, anti-hypertensive, anti-diabetic, anti-inflammatory, anti-HIV, and kinase inhibition [10,11,12,13]. Besides their pharmacological applications, structural modification of these compounds rendered them attractive fluorescent probes, which was vastly exploited by researchers expanding their applications in to optical imaging and sensing [14,15].
2. Structure, Isomerism, and Synthesis of Quinazolines
Quinazoline is a bicyclic heterocyclic compound formed from the fusion of two six-membered aromatic rings of benzene and pyrimidine. These compounds belong to 1,3-benzodiazine (diazonaphthalene) category which contains two nitrogen atoms. Based on the position of nitrogen atoms, the quinazoline ring can exist in four isomeric forms such as quinazoline, quinoxaline, cinnolines, and pthalazines (Figure 1). The oxidized form of quinazolines are called quinazolinones, which can be further classified into 2(1H)-quinazolinones, 4(3H)-quinazolinones, and 2,4(1H,3H)-quinazolinedione based on the position of the keto group (Figure 1) [16].
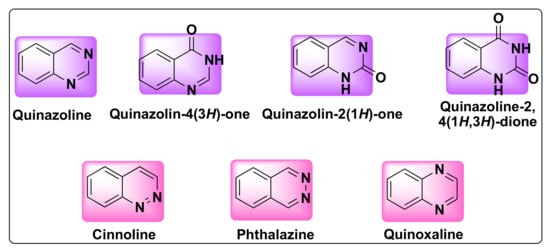
Figure 1. Structures of isomeric forms of quinazolines and its oxidized forms.
The first synthesis of quinazoline was accomplished by Bischler and Lang in the year 1895 [17]. Although these fused ring compounds were known as ‘benzo-1,3-diazine’ in the beginning of their discovery, Weddige later named them as ‘quinazolines’ in the year 1887. The discovery of febrifugine, an anti-malarial drug marked the beginning of an extensive research on the pharmacological activities of quinazolines [18]. The history of quinazoline dates back to 1869, when Griess synthesized 2-cyano-3,4-dihydro-4-oxoquinazoline, the first derivative of quinazoline. Vasicine (peganine), known for its bronchodilatory and anti-tuberculosis activity, was the first quinazoline alkaloid isolated from Justicia adhatoda [19].
Owing to its ubiquitous nature, synthetic versatility and widespread biological and pharmacological applications, numerous synthetic methods have come to light for the facile synthesis of quinazoline and its derivatives both for therapeutic and imaging purposes [15]. The most notable among them is the copper-catalyzed intramolecular N-arylation, Rhodium-catalyzed ortho-amidation, cyclocondensation, tetrabutyl ammonium fluoride promoted cyclization of N-imidoyl-o-alkynylanilines, Niementowski reaction, and benzotrizol-1-yloxytris(dimethylamino) phosphonium hexafluorophosphate (BOP)-mediated ring closures [8,17,20].
3. Quinazolines as Anti-Cancer Agents
Quinazolines are widely acclaimed for their efficiency as anti-cancer agents. Several quinazoline based kinase inhibitors such as afatinib, lapatinib, gefitinib, dacomitinib, tucatinib, and erlotinib are FDA approved for cancer treatment which prove their therapeutic efficiency as anti-tumor agents (Figure 2). Various approaches were proposed to explain the mechanism of their anti-tumor activity, which can be attributed to one or more of the following pathways: (1) Tubulin inhibition, (2) Epidermal growth factor receptor (EGFR) inhibition, (3) Inhibition of DNA damage/repair mechanism, and (4) Inhibition of thymidylate synthetase [21,22].
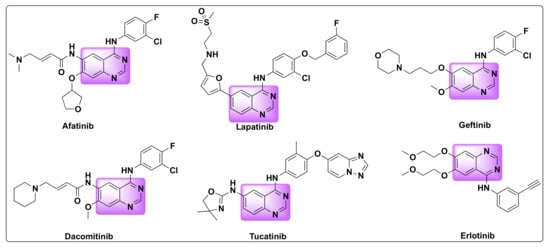
Figure 2. FDA approved quinazoline based drugs for the treatment of lung and breast cancer.
4. Histone Deacetylase Inhibition as Anti-Cancer Agents
Histone deacetylases (HDACs) were discovered by Vincent Allfrey who identified that this set of enzymes can remove acetyl groups from histones [23]. Based on the homology sequence and subcellular localization, HDACs are divided in to four classes—Class I (HDAC-1, 2, 3 and 8), Class II (HDAC-4, 5, 6, 7, 9 and 10), Class III (SERT protein 1–7), and Class IV (HDAC-11). All the HDACs are Zn+2 dependent enzymes except Class III that are NAD+ dependent and not Zn+2 dependent [24]. The fact that these enzymes are overexpressed in several human cancers and are involved in several critical biological events such as DNA damage repair, regulation of transcription, cell cycle, autophagy, and other stress responses besides deacetylation of histone and non-histone residues rendered these substrates as attractive cancer-therapeutic targets [25,26,27]. Therefore, the development of small molecule HDAC inhibitors has gained a lot of attention in the past decade [28,29,30,31]. Currently, there are five FDA approved HDAC inhibitors available for cancer treatment: Panobinostat, vorinostat (SAHA), belinostat (PXD-101), romidepsin (FK-228), and tucidinostat (HBI-8000) (Figure 3).
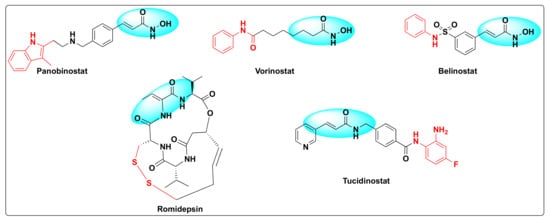
Figure 3. Some selected examples of HDAC inhibitors.
5. Multi-Targeted Therapy and Multi-Drug Resistance
As commonly known, drug resistance is the one of the major challenges encountered in the cancer treatment by chemotherapy [3,32]. Currently, anti-cancer drugs available in the market suffer from two major challenges: toxicity and drug resistance [33]. Based on the time of development, resistance to cancer drugs can be either intrinsic or acquired and can be the outcome of several factors. Some of them include drug metabolism, sequestration of anti-cancer drugs in lysosomes, overexpression of ATP binding cassette (ABC) transporters leading to the extrusion of anti-cancer drugs, alterations in the tumor micro-environment, genetic mutations, drug inactivation, and inhibition of apoptosis just to name a few [34,35,36]. Given the complexity and heterogeneity in the tumor invasion and progression and considering the quick adaptation of cancer drugs to any single chemotherapeutic agent, cocktail or multi-drug therapy which includes treatment with a combination of two or more drugs has become a most common approach in the recent years to treat tumor malignancies, in order to circumvent the drug resistance and improve the safety and efficacy of cancer treatment with less toxicity, multi-targeted therapy has evolved as an effective tool in the field of drug discovery [3,37,38].
However, multi-drug resistance (MDR) is another major hurdle encountered in cancer chemotherapy, which is a phenomenon in which cancer cells develop resistance to structurally or pharmacologically unrelated drugs. Several strategies were developed to overcome the MDR that include inhibition of membrane transport proteins (ABC), gene silencing by turning off the drivers of MDR, transcriptional regulation, targeted delivery of anti-cancer drugs, and hybrid/chimeric drug approaches are some of them [3,32].
This entry is adapted from the peer-reviewed paper 10.3390/molecules27072294
This entry is offline, you can click here to edit this entry!