Caffeine is a natural trimethyl xanthine alkaloid in which the three methyl groups are located at positions 1, 3, and 7 (1,3,7-Trimethylxanthine). Caffeine has high oral bioavailability, with 99% of caffeine being absorbed from the gastrointestinal (GI) tract into the bloodstream 45 min after ingestion. A peak plasma concentration of 1–10 μM (0.25–2 mg/L) reached 15–120 min post oral ingestion from a cup of coffee in humans. Caffeine, a key psychoactive ingredient in coffee, is a short-acting neurostimulator with known neuromodulator effects on the brain by inhibiting phosphodiesterase, mobilizing intracellular calcium, antagonism of adenosine receptors, and modulation of GABA receptor function. Rodent studies have also reported caffeine can inhibit amylogenic-Aβ protein production and improve cognition in rodent AD models. However, results from previous clinical studies were controversial, with some reporting caffeine to be neuroprotective, while others report no effect or even detrimental effects on cognition.
- caffeine
- coffee
- cognition
- Alzheimer’s disease
- dementia
1. Introduction
2. Effect of Coffee/Caffeine
Studies by Haller et al. [14][15] showed in early cognitive decline, there is an increase in compensatory basal activity diffused through the posto-temporal region of the brain, which increases the brain’s sensitivity to the neuroprotective action of caffeine [14][15]. Furthermore, MRI studies by Ritchie et al. [16] and Haller et al. [17] showed that caffeine reduces the amount of white matter lesion/cranial volume in cognitively stable elders, contributing to cognitive decline in Dementia/AD [17][18]. Ritchie et al. [18] also showed increased cerebral perfusion in chronic coffee consumers, indicating a possible neuroprotective mechanism of coffee [18]. Moreover, Gelber et al. [19] found high caffeine levels were associated with a lower-odds of having any brain lesion types at autopsy [19]. However, an epidemiological study by Kim et al. [20] did not find any association between coffee intake and hypometabolism, atrophy of AD signature, and WMH volume; instead, it found that coffee exerted a neuroprotective effect by reducing the levels of Aβ [20].
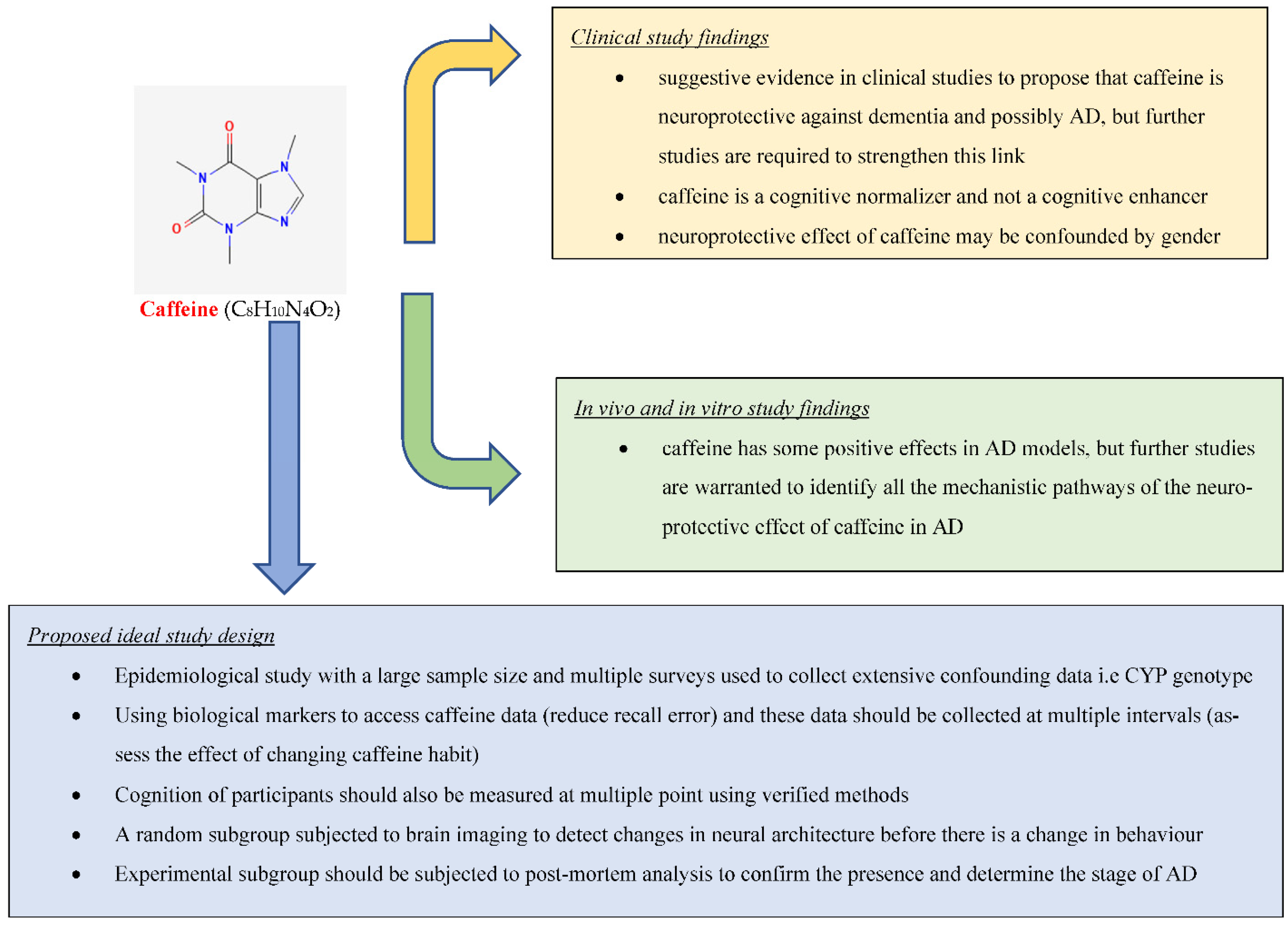
3. Conclusions
This research found suggestive evidence in clinical studies to propose that caffeine is neuroprotective against dementia and possibly AD, but further studies are required to strengthen this link (Figure 1). It also found strong evidence based on in vivo and in vitro studies that caffeine has some positive effects in AD models, but further studies are warranted to identify all the mechanistic pathways of the neuroprotective effect of caffeine in AD.This entry is adapted from the peer-reviewed paper 10.3390/molecules27123737
References
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517.
- Prince, M.J.; Wimo, A.; Guerchet, M.M.; Ali, G.C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015—The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015.
- Karran, E.; Mercken, M.; Strooper, B.D. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712.
- Karch, C.M.; Goate, A.M. Alzheimer’s Disease Risk Genes and Mechanisms of Disease Pathogenesis. Biol. Psychiatry 2015, 77, 43–51.
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, 115–124.
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatry 2011, 16, 903–907.
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794.
- De Bruijn, R.F.; Bos, M.J.; Portegies, M.L.; Hofman, A.; Franco, O.H.; Koudstaal, P.J.; Ikram, M.A. The potential for prevention of dementia across two decades: The prospective, population-based Rotterdam Study. BMC Med. 2015, 13, 132.
- Panza, F.; Solfrizzi, V.; Barulli, M.R.; Bonfiglio, C.; Guerra, V.; Osella, A.; Seripa, D.; Sabba, C.; Pilotto, A.; Logroscino, G. Coffee, tea, and caffeine consumption and prevention of late-life cognitive decline and dementia: A systematic review. J. Nutr. Health Aging 2015, 19, 313–328.
- Kim, J.; Lee, K.W. Coffee and Its Active Compounds Are Neuroprotective; Elsevier: Amsterdam, The Netherlands, 2015; pp. 423–427.
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Kumar, G.; Gedda, M.R.; Tiwari, N.; Patnaik, R.; Singh, R.K.; Singh, S.P. Effect of Chlorogenic Acid Supplementation in MPTP-Intoxicated Mouse. Front. Pharm. 2018, 9, 757.
- Balakrishnan, R.; Azam, S.; Cho, D.-Y.; Su-Kim, I.; Choi, D.-K. Natural Phytochemicals as Novel Therapeutic Strategies to Prevent and Treat Parkinson’s Disease: Current Knowledge and Future Perspectives. Oxidative Med. Cell. Longev. 2021, 2021, 6680935.
- Fahanik-Babaei, J.; Baluchnejadmojarad, T.; Nikbakht, F.; Roghani, M. Trigonelline protects hippocampus against intracerebral Aβ(1–40) as a model of Alzheimer’s disease in the rat: Insights into underlying mechanisms. Metab. Brain Dis. 2019, 34, 191–201.
- Haller, S.; Montandon, M.-L.; Rodriguez, C.; Moser, D.; Toma, S.; Hofmeister, J.; Giannakopoulos, P. Caffeine impact on working memory-related network activation patterns in early stages of cognitive decline. Neuroradiology 2017, 59, 387–395.
- Haller, S.; Montandon, M.-L.; Rodriguez, C.; Moser, D.; Toma, S.; Hofmeister, J.; Sinanaj, I.; Lovblad, K.-O.; Giannakopoulos, P. Acute Caffeine Administration Effect on Brain Activation Patterns in Mild Cognitive Impairment. J. Alzheimers Dis. 2014, 41, 101–112.
- Ritchie, K.; Artero, S.; Portet, F.; Brickman, A.; Muraskin, J.; Beanino, E.; Ancelin, M.-L.; Carriere, I. Caffeine, Cognitive Functioning, and White Matter Lesions in the Elderly: Establishing Causality from Epidemiological Evidence. J. Alzheimers Dis. 2010, 20, S161–S166.
- Haller, S.; Montandon, M.-L.; Rodriguez, C.; Herrmann, F.R.; Giannakopoulos, P. Impact of Coffee, Wine, and Chocolate Consumption on Cognitive Outcome and MRI Parameters in Old Age. Nutrients 2018, 10, 1391.
- Ritchie, K.; Ancelin, M.L.; Amieva, H.; Rouaud, O.; Carriere, I. The association between caffeine and cognitive decline: Examining alternative causal hypotheses. Int. Psychogeriatr. 2014, 26, 581–590.
- Gelber, R.P.; Petrovitch, H.; Masaki, K.H.; Ross, G.W.; White, L.R. Coffee Intake in Midlife and Risk of Dementia and its Neuropathologic Correlates. J. Alzheimers Dis. 2011, 23, 607–615.
- Kim, J.W.; Byun, M.S.; Yi, D.; Lee, J.H.; Jeon, S.Y.; Jung, G.; Lee, H.N.; Sohn, B.K.; Lee, J.-Y.; Kim, Y.K.; et al. Coffee intake and decreased amyloid pathology in human brain. Transl. Psychiatry 2019, 9, 27.
- West, R.K.; Ravona-Springer, R.; Livny, A.; Heymann, A.; Shahar, D.; Leroith, D.; Preiss, R.; Zukran, R.; Silverman, J.M.; Schnaider, M. Age Modulates the Association of Caffeine Intake with Cognition and With Gray Matter in Elderly Diabetics. J. Gerontol. Ser. A-Biol. Sci. Med. Sci. 2019, 74, 683–688.
- Sugiyama, K.; Tomata, Y.; Kaiho, Y.; Honkura, K.; Sugawara, Y.; Tsuji, I. Association between Coffee Consumption and Incident Risk of Disabling Dementia in Elderly Japanese: The Ohsaki Cohort 2006 Study. J. Alzheimers Dis. 2016, 50, 491–500.
- Ritchie, K.; Carriere, I.; de Mendonca, A.; Portet, F.; Dartigues, J.F.; Rouaud, O.; Barberger-Gateau, P.; Ancelin, M.L. The neuroprotective effects of caffeine—A prospective population study (the Three City Study). Neurology 2007, 69, 536–545.
- Iranpour, S.; Saadati, H.M.; Koohi, F.; Sabour, S. Association between caffeine intake and cognitive function in adults; effect modification by sex: Data from National Health and Nutrition Examination Survey (NHANES) 2013–2014. Clin. Nutr. 2020, 39, 2158–2168.
- Janitschke, D.; Nelke, C.; Lauer, A.A.; Regner, L.; Winkler, J.; Thiel, A.; Grimm, H.S.; Hartmann, T.; Grimm, M.O.W. Effect of Caffeine and Other Methylxanthines on A beta-Homeostasis in SH-SY5Y Cells. Biomolecules 2019, 9, 689.
- Arendash, G.W.; Schleif, W.; Rezai-Zadeh, K.; Jackson, E.K.; Zacharia, L.C.; Cracchiolo, J.R.; Shippy, D.; Tan, J. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain β-amyloid production. Neuroscience 2006, 142, 941–952.
- Arendash, G.W.; Mori, T.; Cao, C.; Mamcarz, M.; Runfeldt, M.; Dickson, A.; Rezai-Zadeh, K.; Tane, J.; Citron, B.A.; Lin, X.; et al. Caffeine reverses cognitive impairment and decreases brain amyloid-beta levels in aged Alzheimer’s disease mice. J. Alzheimers Dis. 2009, 17, 661–680.
- Cao, C.; Cirrito, J.R.; Lin, X.; Wang, L.; Verges, D.K.; Dickson, A.; Mamcarz, M.; Zhang, C.; Mori, T.; Arendash, G.W.; et al. Caffeine suppresses amyloid-beta levels in plasma and brain of Alzheimer’s disease transgenic mice. J. Alzheimers Dis. 2009, 17, 681–697.
- Zhao, Z.A.; Zhao, Y.; Ning, Y.L.; Yang, N.; Peng, Y.; Li, P.; Chen, X.Y.; Liu, D.; Wang, H.; Chen, X.; et al. Adenosine A(2A) receptor inactivation alleviates early-onset cognitive dysfunction after traumatic brain injury involving an inhibition of tau hyperphosphorylation. Transl. Psychiatry 2017, 7, e1123.
- Bortolotto, J.W.; de Melo, G.M.; Cognato, G.d.P.; Moreira Vianna, M.R.; Bonan, C.D. Modulation of adenosine signaling prevents scopolamine-induced cognitive impairment in zebrafish. Neurobiol. Learn. Mem. 2015, 118, 113–119.
- Li, S.; Geiger, N.H.; Soliman, M.L.; Hui, L.; Geiger, J.D.; Chen, X. Caffeine, Through Adenosine A(3) Receptor-Mediated Actions, Suppresses Amyloid-beta Protein Precursor Internalization and Amyloid-beta Generation. J. Alzheimers Dis. 2015, 47, 73–83.
- Espinosa, J.; Rocha, A.; Nunes, F.; Costa, M.S.; Schein, V.; Kazlauckas, V.; Kalinine, E.; Souza, D.O.; Cunha, R.A.; Porciuncula, L.O. Caffeine Consumption Prevents Memory Impairment, Neuronal Damage, and Adenosine A(2A) Receptors Upregulation in the Hippocampus of a Rat Model of Sporadic Dementia. J. Alzheimers Dis. 2013, 34, 509–518.
- Dall’Igna, O.P.; Fett, P.; Gomes, M.W.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Caffeine and adenosine A(2a) receptor antagonists prevent beta-amyloid (25-35)-induced cognitive deficits in mice. Exp. Neurol. 2007, 203, 241–245.
- Mancini, R.S.; Wang, Y.; Weaves, D.F. Phenylindanes in Brewed Coffee Inhibit Amyloid-Beta and Tau Aggregation. Front. Neurosci. 2018, 12, 735.
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Müller, C.E.; Buée, L.; et al. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging 2014, 35, 2079–2090.
- Moy, G.A.; McNay, E.C. Caffeine prevents weight gain and cognitive impairment caused by a high-fat diet while elevating hippocampal BDNF. Physiol. Behav. 2013, 109, 69–74.
- Han, K.; Jia, N.; Li, J.; Yang, L.; Min, L.-Q. Chronic caffeine treatment reverses memory impairment and the expression of brain BNDF and TrkB in the PS1/APP double transgenic mouse model of Alzheimer’s disease. Mol. Med. Rep. 2013, 8, 737–740.
- Mohamed, T.; Osman, W.; Tin, G.; Rao, P.P.N. Selective inhibition of human acetylcholinesterase by xanthine derivatives: In vitro inhibition and molecular modeling investigations. Bioorganic Med. Chem. Lett. 2013, 23, 4336–4341.
- Pohanka, M.; Dobes, P. Caffeine Inhibits Acetylcholinesterase, But Not Butyrylcholinesterase. Int. J. Mol. Sci. 2013, 14, 9873–9882.
- Gastaldo, I.P.; Himbert, S.; Ram, U.; Rheinstadter, M.C. The Effects of Resveratrol, Caffeine, beta-Carotene, and Epigallocatechin Gallate (EGCG) on Amyloid-beta(25-35) Aggregation in Synthetic Brain Membranes. Mol. Nutr. Food Res. 2020, 64, 2000632.
- Zappettini, S.; Faivre, E.; Ghestem, A.; Carrier, S.; Buee, L.; Blum, D.; Esclapez, M.; Bernard, C. Caffeine Consumption during Pregnancy Accelerates the Development of Cognitive Deficits in Offspring in a Model of Tauopathy. Front. Cell. Neurosci. 2019, 13, 438.
- Soliman, M.L.; Geiger, J.D.; Chen, X. Caffeine Blocks HIV-1 Tat-Induced Amyloid Beta Production and Tau Phosphorylation. J. Neuroimmune Pharmacol. 2017, 12, 163–170.
- Cao, C.; Wang, L.; Lin, X.; Mamcarz, M.; Zhang, C.; Bai, G.; Nong, J.; Sussman, S.; Arendash, G. Caffeine synergizes with another coffee component to increase plasmato cognitive benefits in Alzheimer’s mice. J. Alzheimers Dis. 2011, 25, 323–335.
- Qosa, H.; Abuznait, A.H.; Hill, R.A.; Kaddoumi, A. Enhanced Brain Amyloid-β Clearance by Rifampicin and Caffeine as a Possible Protective Mechanism Against Alzheimer’s Disease. J. Alzheimers Dis. 2012, 31, 151–165.
- Reznikov, L.R.; Pasumarthi, R.K.; Fadel, J.R. Caffeine elicits c-Fos expression in horizontal diagonal band cholinergic neurons. Neuroreport 2009, 20, 1609–1612.
- Laurent, C.; Burnouf, S.; Ferry, B.; Batalha, V.L.; Coelho, J.E.; Baqi, Y.; Malik, E.; Mariciniak, E.; Parrot, S.; Van der Jeugd, A.; et al. A(2A) adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol. Psychiatry 2016, 21, 97–107.