Gastric cancer is one of the most aggressive tumors in the clinic that is resistant to chemotherapy. Gastric tumors are rich in hypoxic niches, and high expression of hypoxia-inducible factor-1α is associated with poor prognosis. Hypoxia is the principal architect of the topographic heterogeneity in tumors. Hypoxia-inducible factor-1α (HIF-1α) reinforces all hallmarks of cancer and donates cancer cells with more aggressive characteristics at hypoxic niches. HIF-1α potently induces sustained growth factor signaling, angiogenesis, epithelial–mesenchymal transition, and replicative immortality. Hypoxia leads to the selection of cancer cells that evade growth suppressors or apoptotic triggers and deregulates cellular energetics. HIF-1α is also associated with genetic instability, tumor-promoting inflammation, and escape from immunity.
- gastric cancer
- hypoxia
- hypoxia-inducible factor-1α
1. HIF-1α and Hallmarks in Gastric Cancer
1.1. HIF-1α and Sustained Growth Factor Signaling in Gastric Cancer
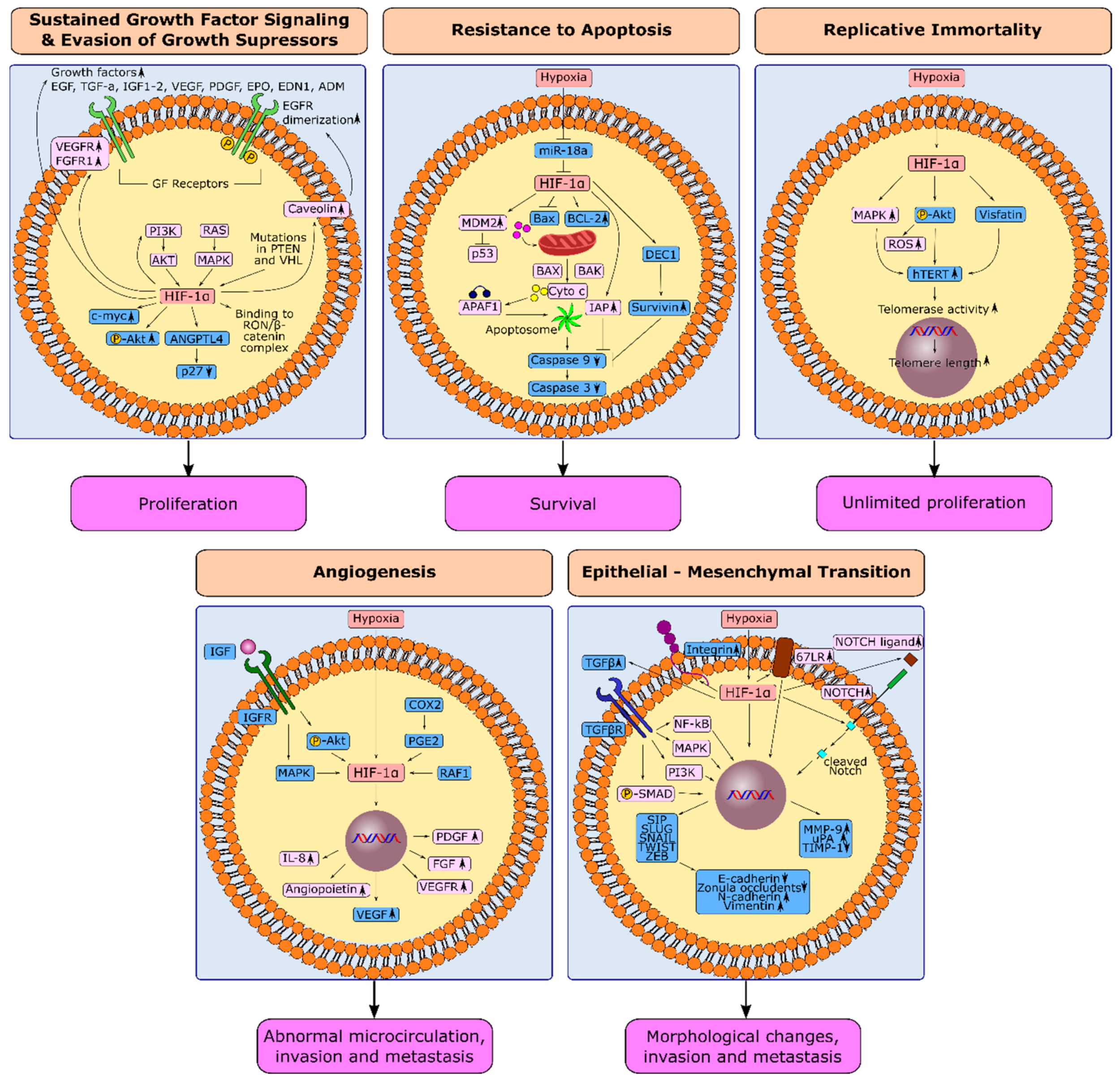
1.2. HIF-1α and Evasion from Growth Suppressors in Gastric Cancer
1.3. HIF-1α and Resistance to Apoptosis in Gastric Cancer
1.4. HIF-1α and Replicative Immortality in Gastric Cancer
1.5. Induction of Angiogenesis through HIF-1α in Gastric Cancer
1.6. Hypoxia-Induced Epithelial-Mesenchymal Transition in Gastric Cancer
2. HIF-1α and Next-Generation Hallmarks in Gastric Cancer
2.1. Hypoxia and Genetic Instability in Gastric Cancer
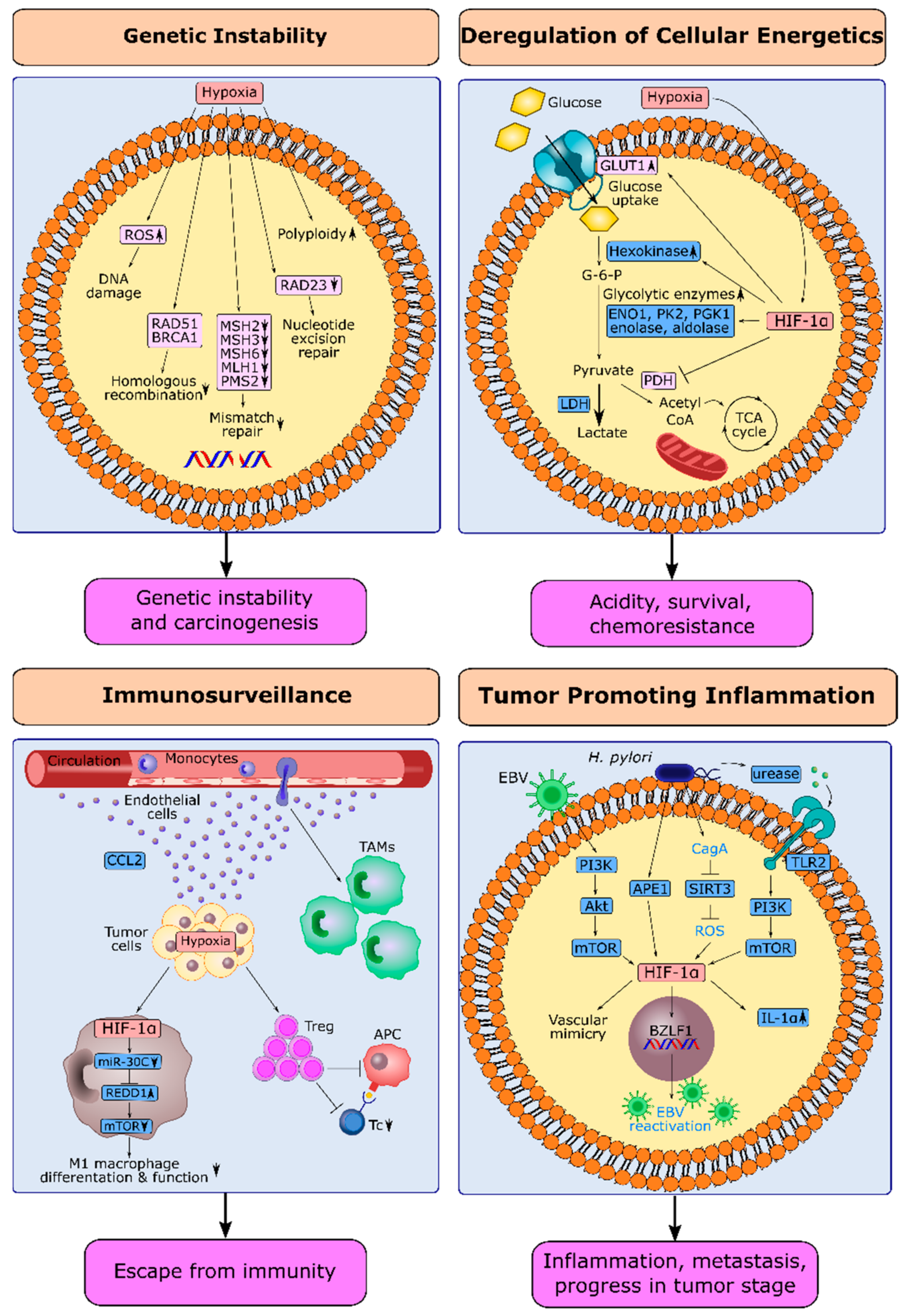
2.2. HIF-1α and Deregulation of Cellular Energetics in Gastric Cancer
2.3. HIF-1α and Escape from Immune Surveillance in Gastric Cancer
2.4. HIF-1α and Tumor-Promoting Inflammation in Gastric Cancer
This entry is adapted from the peer-reviewed paper 10.3390/cancers14112711
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Powis, G.; Kirkpatrick, L. Hypoxia inducible factor-1alpha as a cancer drug target. Mol Cancer Ther. 2014, 3, 647–654.
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214.
- Lu, Y.; Liu, Y.; Oeck, S.; Zhang, G.J.; Schramm, A.; Glazer, P.M. Hypoxia Induces Resistance to EGFR Inhibitors in Lung Cancer Cells via Upregulation of FGFR1 and the MAPK Pathway. Cancer Res. 2020, 80, 4655–4667.
- Waltenberger, J.; Mayr, U.; Pentz, S.; Hombach, V. Functional Upregulation of the Vascular Endothelial Growth Factor Receptor KDR by Hypoxia. Circulation 1996, 94, 1647–1654.
- Wang, Y.; Roche, O.; Xu, C.; Moriyama, E.H.; Heir, P.; Chung, J.; Roos, F.C.; Chen, Y.; Finak, G.; Milosevic, M.; et al. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor–mediated upregulation of caveolin-1. Proc. Natl. Acad. Sci. USA 2012, 109, 4892–4897.
- Kitajima, Y.; Miyazaki, K. The Critical Impact of HIF-1a on Gastric Cancer Biology. Cancers 2013, 5, 15–26.
- Li, X.; Lu, Y.; Liang, K.; Pan, T.; Mendelsohn, J.; Fan, Z. Requirement of hypoxia-inducible factor-1α down-regulation in mediating the antitumor activity of the anti–epidermal growth factor receptor monoclonal antibody cetuximab. Mol. Cancer Ther. 2008, 7, 1207–1217.
- Zhou, D.; Huang, L.; Zhou, Y.; Wei, T.; Yang, L.; Li, C. RON and RONΔ160 promote gastric cancer cell proliferation, migration, and adaption to hypoxia via interaction with β-catenin. Aging 2019, 11, 2735–2748.
- Tamura, G. Alterations of tumor suppressor and tumor-related genes in the development and progression of gastric cancer. World J. Gastroenterol. 2006, 12, 192–198.
- Yang, L.; Kuang, L.G.; Zheng, H.C.; Li, J.Y.; Wu, D.Y.; Zhang, S.M.; Xin, Y.; Chen, Y.; Yang, S. PTEN encoding product: A marker for tumorigenesis and progression of gastric carcinoma. World J. Gastroenterol. 2003, 9, 35–39.
- Zheng, H.-C.; Qiu, Y.-H.; Zhao, S. The roles of PTEN expression in gastric cancer: A bibliometric, meta and bioinformatics analysis. Oncotarget 2018, 9 (Suppl. 1), s1074–s1089.
- Lee, H.S.; Lee, H.K.; Kim, H.S.; Yang, H.K.; Kim, W.H. Tumour suppressor gene expression correlates with gastric cancer prognosis. J. Pathol. 2003, 200, 39–46.
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296.
- Sumiyoshi, Y.; Kakeji, Y.; Egashira, A.; Mizokami, K.; Orita, H.; Maehara, Y. Overexpression of Hypoxia-Inducible Factor 1α and p53 Is a Marker for an Unfavorable Prognosis in Gastric Cancer. Clin. Cancer Res. 2006, 12, 5112–5117.
- Hammond, E.M.; Giaccia, A.J. Hypoxia-Inducible Factor-1 and p53: Friends, Acquaintances, or Strangers? Clin. Cancer Res. 2006, 12, 5007–5009.
- Ruan, K.; Song, G.; Ouyang, G. Role of hypoxia in the hallmarks of human cancer. J. Cell. Biochem. 2009, 107, 1053–1062.
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673.
- Kunz, M.; Ibrahim, S.; Koczan, D.; Thiesen, H.J.; Köhler, H.J.; Acker, T.; Plate, K.H.; Ludwig, S.; Rapp, U.R.; Bröcker, E.B.; et al. Activation of c-Jun NH2-terminal kinase/stress-activated protein kinase (JNK/SAPK) is critical for hypoxia-induced apoptosis of human malignant melanoma. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 2001, 12, 137–145.
- Graeber, T.G.; Osmanian, C.; Jacks, T.; Housman, D.E.; Koch, C.J.; Lowe, S.W.; Giaccia, A.J. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996, 379, 88–91.
- Wang, Y.; Pakunlu, R.I.; Tsao, W.; Pozharov, V.; Minko, T. Bimodal Effect of Hypoxia in Cancer: Role of Hypoxia Inducible Factor in Apoptosis. Mol. Pharm. 2004, 1, 156–165.
- Dong, Z.; Venkatachalam, M.A.; Wang, J.; Patel, Y.; Saikumar, P.; Semenza, G.L.; Force, T.; Nishiyama, J. Up-regulation of Apoptosis Inhibitory Protein IAP-2 by Hypoxia: HIF-1-independent mechanisms. J. Biol. Chem. 2001, 276, 18702–18709.
- Zhang, L.; Hill, R.P. Hypoxia Enhances Metastatic Efficiency by Up-Regulating Mdm2 in KHT Cells and Increasing Resistance to Apoptosis. Cancer Res. 2004, 64, 4180–4189.
- Alvarez-Tejado, M.; Naranjo-Suárez, S.; Jiménez, C.; Carrera, A.C.; Landázuri, M.O.; del Peso, L. Hypoxia Induces the Activation of the Phosphatidylinositol 3-Kinase/Akt Cell Survival Pathway in PC12 Cells: Protective Role in Apoptosis. J. Biol. Chem. 2001, 276, 22368–22374.
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.-Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial Akt Regulation of Hypoxic Tumor Reprogramming. Cancer Cell 2016, 30, 257–272.
- Kim, M.; Park, S.Y.; Pai, H.S.; Kim, T.H.; Billiar, T.R.; Seol, D.W. Hypoxia Inhibits Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Induced Apoptosis by Blocking Bax Translocation. Cancer Res. 2004, 64, 4078–4081.
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014.
- Jia, Y.; Hu, R.; Li, P.; Zheng, Y.; Wang, Y.; Ma, X. DEC1 is required for anti-apoptotic activity of gastric cancer cells under hypoxia by promoting Survivin expression. Gastric Cancer 2018, 21, 632–642.
- Wu, F.; Huang, W.; Wang, X. microRNA-18a regulates gastric carcinoma cell apoptosis and invasion by suppressing hypoxia-inducible factor-1α expression. Exp. Ther. Med. 2015, 10, 717–722.
- Rohwer, N.; Welzel, M.; Daskalow, K.; Pfander, D.; Wiedenmann, B.; Detjen, K.; Cramer, T. Hypoxia-Inducible Factor 1 Mediates Anoikis Resistance via Suppression of 5 Integrin. Cancer Res. 2008, 68, 10113–10120.
- Baba, K.; Kitajima, Y.; Miyake, S.; Nakamura, J.; Wakiyama, K.; Sato, H.; Okuyama, K.; Kitagawa, H.; Tanaka, T.; Hiraki, M.; et al. Hypoxia-induced ANGPTL4 sustains tumour growth and anoikis resistance through different mechanisms in scirrhous gastric cancer cell lines. Sci. Rep. 2017, 7, 11127.
- Shay, J.W.; Wright, W.E. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell Biol. 2000, 1, 72–76.
- Heeg, S. Variations in telomere maintenance and the role of telomerase inhibition in gastrointestinal cancer. Pharmacogenomics Pers. Med. 2015, 8, 171–180.
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183.
- Seimiya, H.; Tanji, M.; Oh-Hara, T.; Tomida, A.; Naasani, I.; Tsuruo, T. Hypoxia Up-Regulates Telomerase Activity via Mitogen-Activated Protein Kinase Signaling in Human Solid Tumor Cells. Biochem. Biophys. Res. Commun. 1999, 260, 365–370.
- Yatabe, N.; Kyo, S.; Maida, Y.; Nishi, H.; Nakamura, M.; Kanaya, T.; Tanaka, M.; Isaka, K.; Ogawa, S.; Inoue, M. HIF-1-mediated activation of telomerase in cervical cancer cells. Oncogene 2004, 23, 3708–3715.
- Lou, F.; Chen, X.; Jalink, M.; Zhu, Q.; Ge, N.; Zhao, S.; Fang, X.; Fan, Y.; Björkholm, M.; Liu, Z.; et al. The Opposing Effect of Hypox-ia-Inducible Factor-2α on Expression of Telomerase Reverse Transcriptase. Mol. Cell. Biol. 2007, 5, 793–800.
- Wang, Y.-P.; Zhu, Z.-Y.; Tang, Y.; Ma, Y.-C. Effects of acute hypoxia on telomere length of rat gastric mucosa tissue and underlying mechanism. Sheng Li Xue Bao 2017, 69, 429–436.
- Mahfouz, N.; Tahtouh, R.; Alaaeddine, N.; EL Hajj, J.; Sarkis, R.; Hachem, R.; Raad, I.; Hilal, G. Gastrointestinal cancer cells treatment with bevacizumab activates a VEGF autoregulatory mechanism involving telomerase catalytic subunit hTERT via PI3K-AKT, HIF-1α and VEGF receptors. PLoS ONE 2017, 12, e0179202.
- Sasaki, T.; Kuniyasu, H.; Luo, Y.; Kitayoshi, M.; Tanabe, E.; Kato, D.; Shinya, S.; Fujii, K.; Ohmori, H.; Yamashita, Y. AKT Activation and Telomerase Reverse Transcriptase Expression are Concurrently Associated with Prognosis of Gastric Cancer. Pathobiology 2014, 81, 36–41.
- Bae, S.-K.; Kim, S.-R.; Kim, J.G.; Kim, J.Y.; Koo, T.H.; Jang, H.-O.; Yun, I.; Yoo, M.-A.; Bae, M.-K. Hypoxic induction of human visfatin gene is directly mediated by hypoxia-inducible factor-1. FEBS Lett. 2006, 580, 4105–4113.
- Segawa, K.; Fukuhara, A.; Hosogai, N.; Morita, K.; Okuno, Y.; Tanaka, M.; Nakagawa, Y.; Kihara, S.; Funahashi, T.; Komuro, R.; et al. Visfatin in adipocytes is upregulated by hypoxia through HIF1α-dependent mechanism. Biochem. Biophys. Res. Commun. 2006, 349, 875–882.
- Mohammadi, M.; Zarghami, N.; Hedayati, M.; Ghaemmaghami, S.; Yamchi, R.M.; Mohaddes, M. Visfatin effects on telomerase gene expression in AGS gastric cancer cell line. Indian J. Cancer 2015, 52, 32–35.
- Lu, G.-W.; Wang, Q.-J.; Xia, M.-M.; Qian, J. Elevated plasma visfatin levels correlate with poor prognosis of gastric cancer patients. Peptides 2014, 58, 60–64.
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133.
- Lv, X.; Li, J.; Zhang, C.; Hu, T.; Li, S.; He, S.; Yan, H.; Tan, Y.; Lei, M.; Wen, M.; et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017, 4, 19–24.
- Urano, N.; Fujiwara, Y.; Doki, Y.; Tsujie, M.; Yamamoto, H.; Miyata, H.; Takiguchi, S.; Yasuda, T.; Yano, M.; Monden, M. Overexpression of hypoxia-inducible factor-1 alpha in gastric adenocarcinoma. Gastric Cancer 2006, 9, 44–49.
- Stoeltzing, O.; McCarty, M.F.; Wey, J.S.; Fan, F.; Liu, W.; Belcheva, A.; Bucana, C.D.; Semenza, G.L.; Ellis, L.M. Role of Hypoxia-Inducible Factor 1α in Gastric Cancer Cell Growth, Angiogenesis, and Vessel Maturation. J. Natl. Cancer Inst. 2004, 96, 946–956.
- Huang, S.-P.; Wu, M.-S.; Shun, C.-T.; Wang, H.-P.; Hsieh, C.-Y.; Kuo, M.-L.; Lin, J.-T. Cyclooxygenase-2 increases hypoxia-inducible factor-1 and vascular endothelial growth factor to promote angiogenesis in gastric carcinoma. J. Biomed. Sci. 2005, 12, 229–241.
- Lee, B.L.; Kim, W.H.; Jung, J.; Cho, S.J.; Park, J.-W.; Kim, J.; Chung, H.-Y.; Chang, M.S.; Nam, S.Y. A hypoxia-independent up-regulation of hypoxia-inducible factor-1 by AKT contributes to angiogenesis in human gastric cancer. Carcinogenesis 2008, 29, 44–51.
- Meng, F.; Ding, J.; Liu, N.; Zhang, J.; Shao, X.; Shen, H.; Xue, Y.; Xie, H.; Fan, D. Inhibition of gastric cancer angiogenesis by vector-based RNA interference for Raf-1. Cancer Biol. Ther. 2005, 4, 120–124.
- Van Beijnum, J.R.; Pieters, W.; Nowak-Sliwinska, P.; Griffioen, A.W. Insulin-like growth factor axis targeting in cancer and tumour angiogenesis-The missing link. Biol. Rev. 2017, 92, 1755–1768.
- Li, H.; Adachi, Y.; Yamamoto, H.; Min, Y.; Ohashi, H.; Ii, M.; Arimura, Y.; Endo, T.; Lee, C.-T.; Carbone, D.P.; et al. Insulin-like growth factor-I receptor blockade reduces tumor angiogenesis and enhances the effects of bevacizumab for a human gastric cancer cell line, MKN45. Cancer 2011, 117, 3135–3147.
- Ru, G.-Q.; Zhao, Z.-S.; Tang, Q.-L.; Xu, W.-J. Expressions of hypoxia inducible factor-1alpha and insulin-like growth factor-II in gastric carcinoma: Correlation with angiogenesis and prognosis. Zhonghua Wai Ke Za Zhi Chin. J. Surg. 2007, 45, 905–908.
- Na Seo, A.; Jung, Y.; Jang, H.; Lee, E.; Bae, H.-I.; Son, T.; Kwon, O.; Chung, H.Y.; Yu, W.; Lee, Y.M. Clinical significance and prognostic role of hypoxia-induced microRNA 382 in gastric adenocarcinoma. PLoS ONE 2019, 14, e0223608.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52.
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691.
- Jiang, J.; Tang, Y.-L.; Liang, X.-H. EMT: A new vision of hypoxia promoting cancer progression. Cancer Biol. Ther. 2011, 11, 714–723.
- Peng, Z.; Wang, C.X.; Fang, E.H.; Wang, G.B.; Tong, Q. Role of epithelial-mesenchymal transition in gastric cancer initiation and progression. World J. Gastroenterol. 2014, 20, 5403–5410.
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428.
- Furuta, C.; Miyamoto, T.; Takagi, T.; Noguchi, Y.; Kaneko, J.; Itoh, S.; Watanabe, T.; Itoh, F. Transforming growth factor-β signaling enhancement by long-term exposure to hypoxia in a tumor microenvironment composed of Lewis lung carcinoma cells. Cancer Sci. 2015, 106, 1524–1533.
- Wendt, M.K.; Allington, T.M.; Schiemann, W.P. Mechanisms of the epithelial–mesenchymal transition by TGF-β. Future Oncol. 2009, 5, 1145–1168.
- Bakin, A.; Rinehart, C.; Tomlinson, A.K.; Arteaga, C.L. p38 mitogen-activated protein kinase is required for TGFβ-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 2002, 115, 3193–3206.
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-β–induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 2007, 178, 437–451.
- Azuma, M.; Motegi, K.; Aota, K.; Yamashita, T.; Yoshida, H.; Sato, M. TGF-β1 Inhibits NF-κB Activity through Induction of IκB-α Expression in Human Salivary Gland Cells: A Possible Mechanism of Growth Suppression by TGF-β1. Exp. Cell Res. 1999, 250, 213–222.
- Jiang, Y.G.; Luo, Y.; He, D.L.; Li, X.; Zhang, L.L.; Peng, T.; Li, M.C.; Lin, Y.H. Role of Wnt/β-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1α. Int. J. Urol. 2007, 14, 1034–1039.
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci USA 2008, 105, 6392–6397.
- Chen, J.; Imanaka, N.; Chen, J.; Griffin, J.D. Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br. J. Cancer 2010, 102, 351–360.
- Matsuoka, J.; Yashiro, M.; Doi, Y.; Fuyuhiro, Y.; Kato, Y.; Shinto, O.; Noda, S.; Kashiwagi, S.; Aomatsu, N.; Hirakawa, T.; et al. Hypoxia Stimulates the EMT of Gastric Cancer Cells through Autocrine TGFβ Signaling. PLoS ONE 2013, 8, e62310.
- Noda, S.; Yashiro, M.; Nshii, T.; Hirakawa, K. Hypoxia upregulates adhesion ability to peritoneum through a transforming growth factor-β-dependent mechanism in diffuse-type gastric cancer cells. Eur. J. Cancer 2010, 46, 995–1005.
- Liu, L.; Sun, L.; Zhao, P.; Yao, L.; Jin, H.; Liang, S.; Wang, Y.; Zhang, D.; Pang, Y.; Shi, Y.; et al. Hypoxia promotes metastasis in human gastric cancer by up-regulating the 67-kDa laminin receptor. Cancer Sci. 2010, 101, 1653–1660.
- Oh, Y.S.; Kim, H.Y.; Song, I.-C.; Yun, H.-J.; Jo, D.-Y.; Kim, S.; Lee, H.J. Hypoxia induces CXCR4 expression and biological activity in gastric cancer cells through activation of hypoxia-inducible factor-1α. Oncol. Rep. 2012, 28, 2239–2246.
- Reynolds, T.Y.; Rockwell, S.; Glazer, P. Genetic instability induced by the tumor microenvironment. Cancer Res. 1996, 56, 5754–5757.
- Bindra, R.S.; Glazer, P.M. Co-repression of mismatch repair gene expression by hypoxia in cancer cells: Role of the Myc/Max network. Cancer Lett. 2007, 252, 93–103.
- Rodríguez-Jiménez, F.J.; Moreno-Manzano, V.; Lucas-Dominguez, R.; Sánchez-Puelles, J.M. Hypoxia Causes Downregulation of Mismatch Repair System and Genomic Instability in Stem Cells. Stem Cells 2008, 26, 2052–2062.
- Yuan, J.; Narayanan, L.; Rockwell, S.; Glazer, P.M. Diminished DNA repair and elevated mutagenesis in mammalian cells exposed to hypoxia and low pH. Cancer Res. 2000, 60, 4372–4376.
- Bindra, R.S.; Glazer, P.M. Genetic instability and the tumor microenvironment: Towards the concept of microenvironment-induced mutagenesis. Mutat. Res. Mol. Mech. Mutagen. 2005, 569, 75–85.
- Koi, M.; Boland, C.R. Tumor hypoxia and genetic alterations in sporadic cancers. J. Obstet. Gynaecol. Res. 2011, 37, 85–98.
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Måseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-Regulation of Rad51 and Decreased Homologous Recombination in Hypoxic Cancer Cells. Mol. Cell. Biol. 2004, 24, 8504–8518.
- Rofstad, E.K.; Johnsen, N.M.; Lyng, H. Hypoxia-induced tetraploidisation of a diploid human melanoma cell line in vitro. Br. J. Cancer Suppl. 1996, 27, S136–S139.
- Lopez-Sánchez, L.M.; Jimenez, C.; Valverde, A.; Hernández, V.; Peñarando, J.; Martinez, A.; López-Pedrera, C.; Muñoz-Castañeda, J.R.; De la Haba-Rodríguez, J.R.; Aranda, E.; et al. CoCl2, a Mimic of Hypoxia, Induces Formation of Polyploid Giant Cells with Stem Characteristics in Colon Cancer. PLoS ONE 2014, 9, e99143.
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033.
- Ghanbari Movahed, Z.G.; Rastegari-Pouyani, M.; Mohammadi, M.; Mansouri, K. Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed. Pharmacother. 2019, 112, 108690.
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350.
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218.
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56.
- Airley, R.E.; Mobasheri, A. Hypoxic Regulation of Glucose Transport, Anaerobic Metabolism and Angiogenesis in Cancer: Novel Pathways and Targets for Anticancer Therapeutics. Chemotherapy 2007, 53, 233–256.
- Hao, L.S.; Liu, Q.; Tian, C.; Zhang, D.X.; Wang, B.; Zhou, D.X.; Li, Z.P.; Yuan, Z.X. Correlation and expression analysis of hypoxia-inducible factor 1α, glucose transporter 1 and lactate dehydrogenase 5 in human gastric cancer. Oncol. Lett. 2019, 18, 1431–1441.
- Jung, J.-H.; Im, S.; Jung, E.S.; Kang, C.S. Clinicopathological Implications of the Expression of Hypoxia-related Proteins in Gastric Cancer. Int. J. Med. Sci. 2013, 10, 1217–1223.
- Semenza, G.L.; Jiang, B.-H.; Leung, S.W.; Passantino, R.; Concordet, J.-P.; Maire, P.; Giallongo, A. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 32529–32537.
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185.
- Song, I.-S.; Wang, A.-G.; Yoon, S.Y.; Kim, J.-M.; Kim, J.H.; Lee, D.-S.; Kim, N.-S. Regulation of glucose metabolism-related genes and VEGF by HIF-1α and HIF-1β, but not HIF-2α, in gastric cancer. Exp. Mol. Med. 2009, 41, 51–58.
- Rho, M.; Kim, J.; Jee, C.D.; Lee, Y.M.; Lee, H.E.; Kim, M.A.; Lee, H.S.; Kim, W.H. Expression of type 2 hexokinase and mitochondria-related genes in gastric carcinoma tissues and cell lines. Anticancer Res. 2007, 27, 251–258.
- Xu, G.; Li, M.; Wu, J.; Qin, C.; Tao, Y.; He, H. Circular RNA circNRIP1 Sponges microRNA-138-5p to Maintain Hypoxia-Induced Resistance to 5-Fluorouracil Through HIF-1α-Dependent Glucose Metabolism in Gastric Carcinoma. Cancer Manag. Res. 2020, 12, 2789–2802.
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579.
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188.
- Lewis, C.; Murdoch, C. Macrophage Responses to Hypoxia: Implications for Tumor Progression and Anti-Cancer Therapies. Am. J. Pathol. 2005, 167, 627–635.
- Tao, L.-L.; Shi, S.-J.; Chen, L.-B.; Huang, G.-C. Expression of monocyte chemotactic protein-1/CCL2 in gastric cancer and its relationship with tumor hypoxia. World J. Gastroenterol. 2014, 20, 4421–4427.
- Zhihua, Y.; Yulin, T.; Yibo, W.; Wei, D.; Yin, C.; Jiahao, X.; Runqiu, J.; Xuezhong, X. Hypoxia decreases macrophage glycolysis and M1 percentage by targeting microRNA-30c and mTOR in human gastric cancer. Cancer Sci. 2019, 110, 2368–2377.
- Ping, W.; Senyan, H.; Li, G.; Yan, C.; Long, L. Increased Lactate in Gastric Cancer Tumor-Infiltrating Lymphocytes Is Related to Impaired T Cell Function Due to miR-34a Deregulated Lactate Dehydrogenase A. Cell. Physiol. Biochem. 2018, 49, 828–836.
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089.
- Deng, B.; Zhu, J.M.; Wang, Y.; Liu, T.T.; Ding, Y.B.; Xiao, W.M.; Lu, G.-T.; Bo, P.; Shen, X.Z. Intratumor Hypoxia Promotes Immune Tolerance by Inducing Regulatory T Cells via TGF-β1 in Gastric Cancer. PLoS ONE 2013, 8, e63777.
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899.
- Baker, K.J.; Houston, A.; Brint, E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front. Immunol. 2019, 10, 1197.
- Xuan, Y.; Wang, Y.N. Hypoxia/IL-1α axis promotes gastric cancer progression and drug resistance. J. Dig. Dis. 2017, 18, 511–520.
- Nishikawa, J.; Iizasa, H.; Yoshiyama, H.; Shimokuri, K.; Kobayashi, Y.; Sasaki, S.; Nakamura, M.; Yanai, H.; Sakai, K.; Suehiro, Y.; et al. Clinical Importance of Epstein–Barr Virus-Associated Gastric Cancer. Cancers 2018, 10, 167.
- Zhang, X.Y.; Zhang, P.Y.; Aboul-Soud, M.A. From inflammation to gastric cancer: Role of Helicobacter pylori. Oncol. Lett. 2017, 13, 543–548.
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209.
- Kraus, R.J.; Yu, X.; Cordes, B.-L.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-inducible factor-1α plays roles in Epstein-Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLOS Pathog. 2017, 13, e1006404.
- Xiang, T.; Lin, Y.-X.; Ma, W.; Zhang, H.-J.; Chen, K.-M.; He, G.-P.; Zhang, X.; Xu, M.; Feng, Q.-S.; Chen, M.-Y.; et al. Vasculogenic mimicry formation in EBV-associated epithelial malignancies. Nat. Commun. 2018, 9, 5009.