Adiponectin is the most abundant endocrine peptide secreted by adipocytes and has widespread physiological activities derived from the combination of the endocrine actions of adipocyte-derived adiponectin with the autocrine or paracrine effects of local adiponectin produced by other cell types, such as skeletal and cardiac myocytes, osteoblasts and ECs. Unlike the majority of adipokines, serum adiponectin levels are reduced in obese individuals [
5] and negatively correlate with chronic subclinical inflammation markers [
21].
Adiponectin is involved in the regulation of energy metabolism and body composition, and serum adiponectin levels are inversely related not only to visceral fat accumulation but also to the grade of insulin resistance (IR) and serum levels of glucose, insulin, and triglycerides (TGs) [
22]. Adiponectin also exerts anti-inflammatory effects in endothelial cells (ECs) [
5], promotes vascular homeostasis by increasing the levels of nitric oxide (NO) [
23], and is involved in the crosstalk between adipose tissue (AT), the immune system and the vascular wall [
24].
Adiponectin secretion is mediated by proliferator-activated receptor gamma (PPARγ), a central regulator of adipocyte biology [
25]. PPARγ is involved in the regulation of lipid metabolism and glucose homeostasis and plays an important role in the cardiovascular system and in vascular development and homeostasis [
26]. Moreover, PPARγ exerts anti-inflammatory action and, accordingly, the transcription of the adiponectin gene in adipocytes is suppressed by pro-inflammatory cytokines, among which is TNF-α [
27].
Adiponectin is found in serum as complexes of different molecular weight: low-molecular-weight (LMW) trimer, medium molecular weight (MMW) hexamer and high-molecular-weight (HMW) multimers that do not interconvert in vivo [
5]. While trimeric and hexameric forms mostly regulate food intake [
28], HMW forms of adiponectin mostly regulate insulin sensitivity, hepatic gluconeogenesis, and other metabolic functions [
29].
The effects of adiponectin are mediated by G-coupled receptors, which occur as two isoforms (AdipoR1 and AdipoR2). These receptors are ubiquitously expressed, AdipoR1 being particularly abundant in skeletal muscle and AdipoR2 in the liver [
30]. Both adiponectin receptors are able to bind the multimerized fragments of adiponectin [
31]. AdipoR1 has a high affinity for the trimer/hexamer forms, while the AdipoR2 preferentially binds to multimers [
32]. Moreover, the adiponectin hexamer and multimer forms bind T-Cadherin, a membrane glycoprotein that can sequester adiponectin on the cell surface and is expressed mainly by endothelial and smooth muscle cells [
33,
34,
35]. Of interest, it is after the binding of adiponectin to T-cadherin that ECs are stimulated to release microvesicles [
36], thus emphasizing the complexity of the crosstalk between ECs and adipocytes [
37].
AdipoR1 and AdipoR2 have also intrinsic ceramidase activity that is greatly amplified by the binding of adiponectin to its receptors and may be the underlying mechanism explaining many of adiponectin-related phenotypes [
38]. This enzyme cleaves fatty acids from ceramides, producing sphingosine, which, in turn, is phosphorylated by a sphingosine kinase to form sphingosine-1-phosphate (S1P). S1P is a pleiotropic lipid mediator that regulates several cell functions via high-affinity G protein-coupled receptors [
38,
39,
40]. Adiponectin lowers hepatic ceramide content through enhanced ceramide catabolism with consequent production of S1P. Accordingly, in obesity, ceramide accumulates in various tissues, partly because of lower amounts of adiponectin [
41].
Adiponectin activates multiple signaling pathways, which mediate its metabolic actions and immunomodulatory effects. The binding of adiponectin to AdipoR1 and R2, through the recruitment of the adaptor protein phosphotyrosine interacting with PH domain and leucine zipper 1 (APPL1), triggers a series of tissue-dependent signal transduction events, including AMP-activated protein kinase (AMPK), p38 mitogen-activated protein kinases (p38 MAPK), protein kinase A (PKA), peroxisome proliferator-activated receptor-α (PPARα), phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), insulin receptor substrate proteins 1 and 2 (IRS1/2)/protein kinase B (Akt) [
5,
42,
43,
44,
45] (
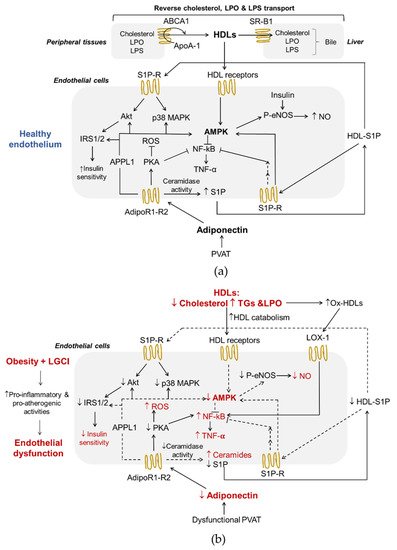
Figure 1a).
Figure 1. Adiponectin and HDLs interplay in endothelial function (a) and obesity-induced endothelial dysfunction (b). Both HDLs and adiponectin contribute to healthy endothelium in physiological conditions.
Adiponectin plays a metabolic role in maintaining energy homeostasis acting through phosphorylation and activation of AMPK [
46,
47]. AMPK is a metabolic sensor that is activated when ATP levels in the cells decrease. Its signaling regulates energy metabolism homeostasis [
48] and can inhibit the inflammatory responses induced by the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) system [
49]. In ECs, the AMPK pathway improves cell function through the activation of endothelial nitric oxide synthase (eNOS) and inhibits the secretion of inflammatory mediators (
Figure 1a). In addition, the activation of protein kinase A (PKA) contributes to promoting NO production and suppresses reactive oxygen species (ROS) generation and NF-κB signaling [
50] (
Figure 1a). In the liver, AMPK activation coordinates the partitioning of fatty acids between oxidative and biosynthetic pathways by increasing fatty acid oxidation capacity and inhibiting de novo lipogenesis [
51], and hinders/blocks enzymes involved in gluconeogenesis promoting a reduction in blood glucose levels [
52]. Moreover, the sequential activation of AMPK, p38 MAPK, and PPARα increase the expression of enzymes involved in fatty acid oxidation [
53]. p38 MAPK serves as a nexus for signal transduction and plays a vital role in numerous biological processes including the production of pro-inflammatory cytokines, such as IL-1β, TNF-α, and IL-6 [
54]. PPARα is a ligand-activated nuclear receptor highly expressed in the liver that acts as a nutritional sensor and grants the adaptation of the rates of fatty acid catabolism, lipogenesis, and ketone body synthesis, in response to feeding and starvation.
Insulin is a well-known regulator of glucose, protein, and lipid metabolism. In addition, insulin promotes NO synthesis via eNOS [
55] (
Figure 1a). The binding of insulin to its receptors induces structural changes due to the auto-phosphorylation of tyrosine residues, followed by downstream events, such as the recruitment of different adaptor proteins (IRS1/2). Different types of insulin-dependent kinases, including Akt, AMPK, and glycogen synthase kinase 3 (GSK-3), can phosphorylate and activate the IRS1/2 [
56].
4. High-Density Lipoproteins
HDLs are a heterogeneous and complex class of lipoproteins with density ranging from 1.063–1.210 g/mL, considerable differences in size, shape, composition and function, produced mainly by the liver and, to a lesser extent, by the small intestine. In human plasma, the large, less dense (1.063–1.125 g/mL) lipid-enriched HDL2 and the small, dense (1.125–1.210 g/mL) protein-enriched HDL3 represent the two major sub-classes of HDLs [
57]. HDLs contain several apolipoproteins (Apos) of which ApoA-I is quantitatively the most relevant and characterizes this lipoprotein class. Other Apos are ApoA-II, ApoA-IV, ApoC-I, ApoC-II, ApoC-III, ApoC-IV, ApoD, ApoE, ApoF, ApoH, ApoJ, ApoL-I and ApoM [
57]. In addition, several enzymes circulate in the bloodstream associated with HDLs, including enzymes involved in lipoprotein remodeling (lecithin-cholesterol acyltransferase, LCAT, cholesterol ester transfer protein, cholesteryl ester transfer protein CETP, and phospholipid transfer protein, PLTP), paraoxonase-1 (PON-1) and lipopolysaccharide (LPS)-binding protein (LBP) [
58]. The main lipids of HDLs are phospholipids (PLs) of which phosphatidylcholine (PC) and sphingomyelin (SM) are the main glycerophospholipids and sphingolipids, respectively. PLs modulate HDLs functions and are the precursors of a variety of regulatory molecules, including lysophospholipids and ceramides. In addition, S1P is transported in circulatory and interstitial fluids by HDLs-bound ApoM.
There are several interactions between HDLs and the endothelium (
Figure 1a). First of all, reverse cholesterol transport (RCT), the ability to transport cholesterol from peripheral tissues back to the liver for excretion in the bile, is the best-known function of HDLs and a process that plays a central role in preventing endothelial dysfunction and atherosclerosis. Of interest, ECs express HDLs’ scavenger receptor B type I (SR-BI), the ATP-binding cassette transporters A1 and G1 (ABCA1 and ABCG1), and the ecto-F1-ATPase [
59]. As shown in
Figure 1a, upon the binding of HDLs to their receptors as well as to S1P receptors, various kinases, including Src, AMPK, p38 MAPK, PI3K and Akt, are activated [
60,
61]. As a result, HDLs enhance endothelial barrier-function and exert anti-inflammatory, anti-apoptotic and anti-adhesive properties [
57,
62,
63,
64]. In addition, HDLs reduce the cellular production of superoxide, an inactivator of the vasodilator NO, by decreasing the activity of endothelial nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [
65], thus preventing ED.
RCT begins with the formation of nascent HDLs particles, which consist mainly of ApoA-I (
Figure 1a). Cholesterol efflux is mediated by the transporters ABCA1, ABCG1 and SR-B1. This step involves the interaction between lipid-free or lipid-free monomeric ApoA-I and ABCA1, while ABCG1 mediates the outflow of cellular cholesterol to lipidated HDL particles. The expression of the ABCA1 and ABCG1 genes is regulated at the transcriptional level by the liver X receptors (LXRs)-α and β [
66]. Like ABCG1, SR-B1 in peripheral cells may also promote cholesterol outflow to mature HDLs particles, but its role in the RCT pathway is particularly important in the liver where it mediates the selective uptake of cholesteryl esters from HDLs [
24]. HDLs also influence triglyceridemia because of their regulatory role on HL activity. HL binds to proteoglycans on the cell surface of hepatocytes and hepatic ECs. HDLs bind to HL and release the enzyme into the circulation where it hydrolyses TGs and PLs of plasma lipoproteins [
67]. HDL2 is more effective in displacing proteoglycan-bound HL than HDL3. In addition, in vivo, and in vitro models suggest that HDLs promote an increase in adiponectin production from AT in a P13K-dependent manner [
68].
Another well-known function of HDLs is their role as anti-inflammatory regulators exerted through interactions with both the vascular endothelium and circulating inflammatory cells [
69]. As mentioned above, HDLs reduce the expression of endothelial adhesion molecules in response to inflammatory mediators and the migration of monocytes into the vascular wall, simultaneously exploiting their antioxidant activities [
57]. HDLs prevent the induction of endothelial 32-kDa putative cysteine protease (CPP32)-like protease, resulting in a decrease in the activity of TNF-α, and, consequently, reduce the apoptotic rate of these cells [
70]. Moreover, HDLs participate in a mechanism of intercellular communication involving the transport and delivery of specific microRNAs (miRNAs), small non-coding RNAs that post-transcriptionally regulate gene expression through translational inhibition and mRNA destabilization [
71]. It has been shown that the transfer of miRNA-223 from HDLs into ECs reduces inflammation by suppressing the expression of intercellular adhesion molecule 1 (ICAM-1) [
72].
The anti-inflammatory properties of HDLs may be due also to their ability to neutralize bacterial products, such as LPS (
Figure 1a). LPS is a bacterial endotoxin with powerful pro-inflammatory activity that can reach the systemic circulation even during the absorption of nutrients in much smaller quantities than those associated with a bacterial infection, but sufficient to contribute to LGCI [
73]. Moreover, LPS decreases HDL cholesterol (HDL-C) and adiponectin levels in vivo [
68] and directly and indirectly participates in the inflammatory reaction in AT during obesity [
74]. The exposure of ECs to this endotoxin results in endothelial activation and production of various pro-inflammatory mediators, and, ultimately, in cellular injury [
75]. Interestingly, very recently, Han et al. have demonstrated that intestine-derived HDL3 traverses the portal vein complexed with LPS-binding protein preventing LPS activation of liver macrophages and supporting extracellular inactivation of this endotoxin [
58].
Finally, the antioxidant activities of HDLs prevent ED via endothelial ABCG1-mediated efflux of cholesterol and 7-oxysterols [
76] and the inhibition of lipid peroxide accumulation because of PON-1 activity. HDLs are the major carrier in the circulation of PON-1, an esterase characterized by three enzymatic activities (lactonase, arylesterase and paraoxonase) that is involved in drug metabolism, and possesses antioxidant and anti-inflammatory properties [
77]. The esterase activities of PON-1 allow the removal of peroxidized fatty acids from PLs, limiting damage resulting from oxidative stress. PON-1 hydrolyses also lactones, including homocysteine thiolactone, a toxic metabolite of homocysteine, which, by modifying protein lysine residues, leads to cell death, altered vessel structure, chronic inflammation, autoimmune response and atherosclerosis [
78]. This PON-1 activity is probably involved in the mechanisms by which HDLs activate eNOS in an inflammatory environment [
60]. Another important antioxidant activity of HDLs can be ascribed to their reverse transport of lipid peroxides [
79] (
Figure 1a). In fact, HDLs can acquire lipid peroxides from low-density lipoproteins (LDLs) and cell membranes holding them in an environment where they may be safely hydrolyzed and from which they may be released to the liver for elimination.
6. Interplay between Adiponectin and HDLs in Endothelial Function and Obesity-Associated ED
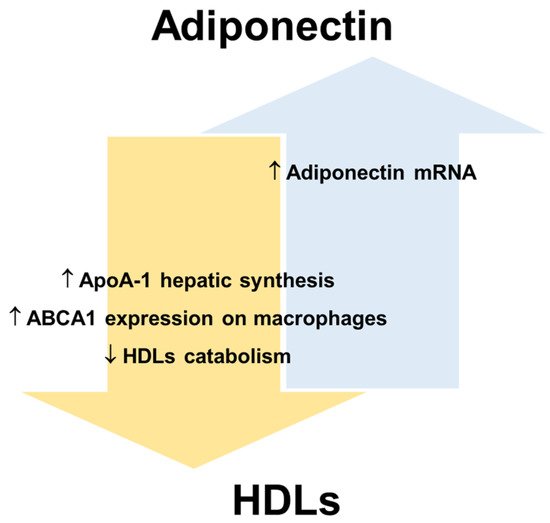
HDLs and adiponectin reciprocally regulate their levels and metabolism (
Figure 3). HDLs can enhance circulating adiponectin levels [
68,
108], while the amount of circulating adiponectin is an independent predictor of cellular cholesterol efflux capacity in humans [
24]. Dias et al. [
109] showed that elevated adiponectin levels are associated with a lower reduction in HDLs function assessed by measuring ApoA-I levels, particle size, cholesterol content and antioxidant capacity in T2DM patients. Nonetheless, the mechanisms that link the metabolism of adiponectin and lipoproteins have not been fully elucidated mainly because analytical difficulties complicate data collection and interpretation.
Figure 3. Reciprocal regulation of adiponectin and HDLs. Apolipoprotein A-1, ApoA-1; ATP-binding cassette transporters A1, ABCA1; high-density lipoproteins, HDLs.
In this regard, human and animal studies suggest that adiponectin promotes not only cellular cholesterol efflux but also HDLs biogenesis [
24], while reducing the catabolism of HDLs/ApoA-I and the clearance rate of VLDLs [
24,
110]. Several human studies have shown that the levels of adiponectin and HDL-C are positively correlated in serum (revised in [
108]). Marsche et al. observed in adult obese subjects a positive correlation between reduced plasma adiponectin and cholesterol efflux capacity of HDLs, independently of sex, BMI, fat distribution, blood pressure, and kidney and liver functions [
111]. A study [
112] shows that the levels of HDL-C significantly improve in obese individuals after bariatric surgery, which determines a rapid reduction of AT and consequently an increase in adiponectin production. In particular, the data show that bariatric surgery promotes an improvement of ApoA-I and adiponectin levels in parallel with an amelioration of cholesterol efflux capacity. For this reason, the increase in adiponectin production, as a crucial modulator of ApoA-I synthesis and hence of HDLs function, may represent a mechanism for decreasing cardiovascular risk associated with obesity [
112]. HDL-C itself is considered a positive factor for endothelial health and is inversely related to CVD risk. Results from human studies suggest that low plasma adiponectin levels are associated with decreased LPL mass and activity [
113,
114]. LPL is a lipolytic enzyme that hydrolyses the TGs of VLDLs and chylomicrons and a decrease in its activity is associated with an increase in plasma TGs and a decrease in HDL-C levels [
115]. Therefore, adiponectin could indirectly maintain HDLs number and function by preventing the enrichment of HDLs in TGs. Importantly, an increase in TGs-rich lipoproteins also leads to ED by dysregulating the cytokine network and decreasing insulin-induced NO synthesis [
116].
Another possible interaction could concern the regulation of ceramide concentration in EC membranes and the production and transport of S1P. HDLs transport significant amounts of ceramides. However, how ceramides are incorporated into HDLs particles is still unknown. It has been hypothesized that PLTP and CETP might transfer ceramides from ApoB-lipoproteins (where ceramides are incorporated during VLDLs assembly) to HDLs and/or that HDLs might directly accept ceramides from plasma membranes [
117]. On the other hand, the ceramidase activity of AdipoRs and the activation of neutral ceramidase by adiponectin decreases ceramide concentration in the membranes of ECs and produces S1P, which also mediates angiogenesis, induces the formation of tight junctions between ECs and plays an important role in maintaining the endothelial barrier. Moreover, S1P can be released into the blood, thus contributing to the pool of circulating S1P [
118,
119,
120,
121]. In addition, the binding of adiponectin to T-cadherin triggers the formation and release of ceramide-containing exosomes, thus lowering EC ceramide content [
122]. Therefore, vascular endothelium is a significant source of circulating S1P because of adiponectin activity and HDLs are an important carrier of this bioactive lipid in serum. LGCI leads to increased production of ceramides by the activation of sphingomyelinase and the accumulation of ceramides in ECs causes ROS overproduction and ED [
23].
In hyperglycemic and dyslipidemic subjects, significantly higher levels of oxidative stress and CVDs risk markers are observed than in healthy subjects, concomitantly with significantly lower levels of adiponectin [
123].
Both chronic inflammation and oxidative stress promote peroxidative damages of proteins and lipids in HDLs resulting in the formation of oxidized HDLs (ox-HDLs) characterized by a loss of antioxidant and anti-inflammatory activities, reduced capacity to remove cholesterol from macrophages and other pro-atherosclerotic effects with negative repercussions also on endothelial function [
124,
125,
126,
127]. The peroxidation of HDL-polyunsaturated fatty acids results in the formation of oxidized lipids and peroxidation end products, including various bioactive aldehydes, such as malondialdehyde (MDA), 4-hydroxynonenal (4-HNE) and acrolein [
64]. These aldehydes are known to contribute to numerous pathologies through the alteration of proteomic, genomic, cellular signaling and metabolic processes [
128] and to be able to modify apolipoproteins, making atherogenic the LDLs [
129] and the HDLs less effective in counteracting atherogenesis [
128]. Moreover, ox-HDLs, like ox-LDLs, induce an increase in the concentration of the oxidized receptor of low-density lipoproteins-1 (LOX-1) in the plasma membrane of ECs [
20] (
Figure 1b). LOX-1 activation by ox-LDLs/HDLs and other ligands causes ED by activating NF-κB and the subsequent induction of adhesion molecules and endothelial apoptosis [
130], and triggering endothelial PKCβII activation, which in turn inhibits eNOS-activating pathways and, consequently, eNOS-dependent NO production [
131] (
Figure 1b). It has been shown that adiponectin selectively binds and inhibits the uptake of oxidized LDLs (oxLDLs) but not of native LDLs by LOX-1 [
132]. Unfortunately, studies have not been published that demonstrate this effect also on ox-HDLs. However, subjects with morbid obesity show an increased level of circulating ox-HDLs, a markedly reduced amount of adiponectin and an increased level of circulating ECs, reliable indicators of vascular injury and damage [
133]. Coherently, the number of endothelial progenitor cell colonies was reduced in these patients, suggesting the presence of an early endothelial stem cell dysfunction and a decreased endothelium repair capacity [
134].
Finally, ox-HDLs significantly contribute to the trans-differentiation of vascular smooth muscle cells (VSMCs) into osteoblasts by enhancing the activity of alkaline phosphatase and calcium deposition, thus promoting vascular calcification and atherosclerosis plaques progression [
135]. Interestingly, adiponectin contributes to the anti-atherogenic phenotype also by counteracting vascular calcification. In particular, some data show that adiponectin reduces calcium deposition in human VSMCs by preventing ox-HDLs-related production of IL-6, WNT-5a and NF-ĸβ (p65) [
136]. Moreover, adiponectin reduces TNF-α -induced calcification in VSMCs through the activation of AMPK [
137] and prevents the osteogenic differentiation of VSMCs by downregulating the expression of the osteogenic transcription factor Osterix through the inhibition of STAT3 phosphorylation and nuclear transport [
138].