Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
Neurodegenerative diseases (NDDs) are disorders that affect both the central and peripheral nervous systems. To name a few causes, NDDs can be caused by ischemia, oxidative and endoplasmic reticulum (ER) cell stress, inflammation, abnormal protein deposition in neural tissue, autoimmune-mediated neuron loss, and viral or prion infections.
- neurological disorder
- neuroinflammation
- proteinopathies
1. Neurodegeneration, Inflammation, and Tumorigenesis in the Central Nervous System
Although the pathogenic function and basic molecular processes underpinning neurodegeneration are complicated, involving genetic, environmental, and endogenous variables linked with aging, their pathogenic function and basic molecular mechanisms are unknown [30,31]. Now, NDDs are categorized based on their known genetic pathways and the primary chemicals found in their protein deposits. These disorders are called ‘protein misfolding’ diseases or proteinopathies because significant structural abnormalities cause them in proteins [32,33]. Numerous fundamental mechanisms underlying neurodegeneration may be initiated at various stages of the neurodegenerative cascade by inflammatory cells and mediators.
Apoptosis: Apoptosis is a form of planned cell death controlled by caspases [34,35,36,37]. It is characterized by the production of membrane-encased apoptotic bodies that are rapidly phagocytozed by macrophages or neighboring cells. Although evidence of apoptotic pathways has been found in animal models of a variety of neurodegenerative illnesses, there is less evidence in human tissues. In Huntington’s disease (HD) models, activation of caspase-1, -3, -8, and -9 as well as cytochrome c release were found in human striatal brain tissue. Similarly, in amyotrophic lateral sclerosis (ALS) and HIV-associated neurodegeneration, caspase activation and neuronal death have been observed [38].
Necroptosis: Necroptosis is a type of programmed cell death defined by the loss of plasma membrane integrity and occurs in the absence of caspase activation. The receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and mixed-lineage kinase domain-like are the two key effector proteins in necroptosis (MLKL). TNF-α, FasL-, and TRAIL are released by astrocytes and can cause necroptosis by activating RIPK1 and MLKL, as shown in ALS mice models [39]. Axonal disease caused by RIPK1 was detected in pathological specimens from ALS patients [40]. In MS pathology samples, necroptotic pathways were also detected [41].
Autophagy, also known as type II programmed cell death, is defined by the buildup of autophagic vacuoles during cell death, along with potentially harmful components such as proteins or damaged organelles. Excessive autophagy can result in cell death and self-destruction. Autophagosomes were found in AD, HD, and PD patients’ damaged neurons. Numerous other triggers, such as food deprivation, mitochondrial toxins, hypoxia, and oxidative stress, can cause autophagy [42,43].
Axonal damage or transection can result in retrograde degeneration of the proximal neuronal cell body, which is associated with a range of degenerative alterations within the cell body, including apoptosis and neuronal perikaryon chromatolysis. Because of the relationship between neuronal apoptosis and axonal damage, inflammation-induced axotomy may result in retrograde (secondary) apoptosis of neuronal cell bodies [44].
Astrogliopathy: Astrogliopathy is a broad word that refers to astrocyte dysfunction. The abnormal buildup of inappropriately phosphorylated tau protein in astrocytes seen in AD, frontal temporal lobe dementia (FTLD), and corticobasal degeneration is referred to as aging-related tau astrogliopathy (ARTAG) [45,46]. Optic neuritis and myelitis characterize neuromyelitis optica (NMO), which can mimic MS. Antibodies against aquaporin-4 (AQP4), which binds to astrocyte water channels, are linked to NMO. NMO is characterized pathologically by a significant loss of immunoreactivity for the astrocytic proteins AQP4 and glial fibrillary acidic protein (GFAP), perivascular deposition of immunoglobulins, and complement activation, even in lesions containing some myelin [47].
Inflammation begins when the body’s immune cells start inflammatory cascades to avoid tissue damage caused by injury or invading pathogens. If the inflammatory response is successful, it eliminates pathogens, initiates wound healing and angiogenesis, and eventually decreases. When the neuroinflammatory response is acute, it is required and even helpful for the neuronal environment, as it aids in pathogen elimination and brain restoration. When serious threats to the neural environment, such as protein aggregates (Lewy bodies, neurofibrillary tangles), build in the brain and sustain inflammation for an extended length of time, continuous gliosis and apoptosis can occur as a result of uncontrolled inflammatory cytokine production. Chronic inflammation is associated to nearly all neurological diseases, including AD, PD, and ALS, as a result of persistent activation [48,49]. In contrast to this protective homeostatic mechanism, inflammation has been implicated in a wide variety of diseases. In recent years, its impact on neurological disorders has been hypothesized as a crucial role in disease progression. Microglia in the CNS form phagocytic morphologies and secrete pro-inflammatory cytokines to interact with astrocytes and neurons. This can result in neurodegeneration, synaptic phagocytosis, reduced neuronal function, microglial activation, inflammatory cytokine release, and even more microglial activation until the neural environment is no longer threatened. Astrocytes are also activated during the inflammatory process, a process known as astrogliosis. Aging is a significant risk factor for neurodegeneration [50,51,52]. In general, older adults have dysregulated cytokine expression (i.e., increased synthesis of pro-inflammatory cytokines and decreased availability of anti-inflammatory cytokines), resulting in a chronic low-grade inflammatory state. Inflammaging is a term that refers to this type of auto-inflammatory disorder that occurs throughout aging [53].
Aside from blood and lymphatic vessels, data suggests that neurogenesis (the growth of new neurons) and axonogenesis (tumor-induced neural sprouting toward the tumor microenvironment) are important in carcinogenesis and cancer progression. Neurogenesis and axonogenesis have been seen in pre-neoplastic lesions, implying that they play a role in the onset of cancer as an early occurrence in the pre-malignant phase [54].
2. New Potential Biomarkers
In contrast to medical symptoms, which are simply indications of a patient’s health as expressed and perceived by the patient, biomarkers, or “biological markers“. In 1998, the Biomarkers Definitions Working Group of the National Institutes of Health defined biomarkers as “evidence of any biological, pathogenic, or pharmacogenomic response to any therapy modification” [115]. Biological markers are any substances, structures, or processes that may be measured inside or outside the body and can influence any changes in the body or the chance of disease prevalence of neurological diseases [116].
2.1. Alzheimer’s Disease
AD is a slowly progressing neurological disease for which there is now no effective cure. Deposition of 42-amino-acid-long amyloid (Aβ) protein in extracellular plaques in the brain is the earliest identifiable disease, which occurs decades before clinical symptoms appear [117]. According to biomarker studies, a buildup is connected to synaptic dysfunction and increased tau phosphorylation and secretion, a microtubule-binding axonal protein abundantly produced in cortical neurons [118]. This dysregulated tau metabolism puts neurons at an elevated risk of degeneration, as intraneuronal neurofibrillary tangles formed of hyperphosphorylated and shortened tau proteins form. Neurodegeneration finally manifests as the AD clinical syndrome, characterized by progressive cognitive deficits [101,119,120]. The pathology of AD is shown in Figure 2.
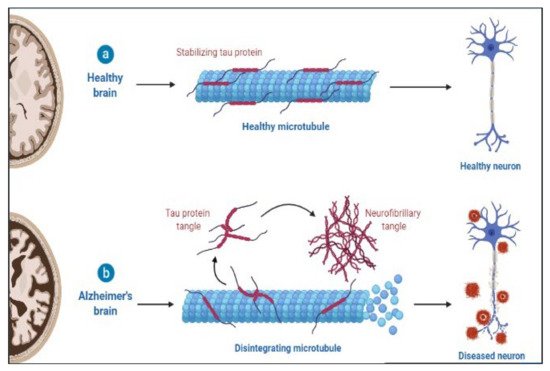
Figure 2. Pathology of AD.
2.2. Aβ Pathology Biomarkers
Extracellular deposition of Aβ, formed by BACE1 and γ-secretase cleavage of amyloid precursor protein (APP), forming plaques is a significant pathological characteristic of AD. It has been hypothesized to represent the primary pathogenic event in the illness [124]. Aβ42 is an APP breakdown product generally transported from the brain interstitial fluid to the CSF and blood via the glymphatic system [125]. Amyloid positron emission tomography (PET) has been validated in comparison with neuropathology, has undergone substantial standardization in terms of quantifying pathology and defining abnormality cut-points, and has adequate usage criteria [126,127,128].
2.3. Tau Pathology Biomarkers
A fundamental pathogenic hallmark of AD is the aggregation of hyperphosphorylated tau in the neuronal soma, generating neurofibrillary tangles. However, tau inclusions in neurons or glial cells are also observed in other neurodegenerative dementias [129]. Together with the CSF Aβ42/Aβ40 ratio, the cornerstone markers totaled tau (T-tau) and phosphorylated tau (P-tau) have been proposed as biomarkers for biologically defining AD and are considered diagnostic in the research criteria for AD [130,131]. Both T-tau and P-tau concentrations in the cerebrospinal fluid (CSF) reflect AD pathogenesis in all neurodegenerative dementias [132]. The most likely explanation is that the higher tau levels in the CSF result from enhanced tau phosphorylation and release by neurons in response to Aβ exposure [133,134].
2.4. Multiple Sclerosis
MS is a chronic autoimmune disease that causes demyelination of the central nervous system and neurodegeneration [135]. Through interplay with immune cells, energy metabolic problems and endocrine abnormalities have been demonstrated to begin MS [136]. Furthermore, viral infection and environmental pollutants have been demonstrated to impair immunological tolerance and trigger the release of proinflammatory factors such as IL-6 and NF-kB in hereditarily vulnerable people [137]. The disease is heterogeneous in radiological and histological alterations, clinical presentation and development, and response to therapy [138,139]. P-gp expression is also increased, which promotes CD4+ and CD8+ T cell migration and amplifies neuroinflammation [140]. T-cells and B-cells mediate inflammatory responses by secreting cytokines that activate inflammatory cells such as microglia behind the BBB [141]. The pathology of MS is shown in Figure 3.
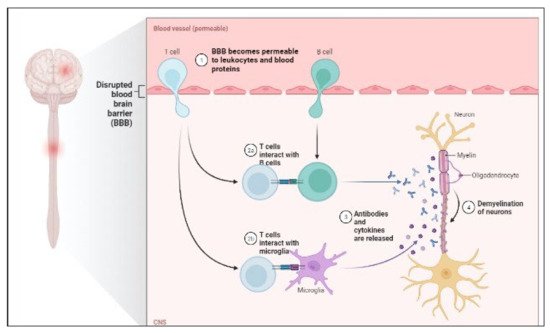
Figure 3. Pathology of MS.
When a patient’s blood serum and CSF fluid are analyzed at the same time, oligoclonal bands are discovered. It has long been known that oligoclonal bands (OCB) can be found in the CSF of MS patients (by isoelectric focusing). Plasma cells in the CNS use immunoglobulin G (IgG) and M to create them (IgM) [142].
After a period in which OCB were not employed for diagnosis according to the McDonald criteria, they have been reintroduced into the diagnostic algorithm in the 2017 update. This shift toward substituting a positive CSF result for dissemination in time rather than in space is pragmatic. However, it underlines clinical neurologists’ responsibility to obtain cutting-edge CSF tests. MS is the most likely diagnosis for patients with typical clinical presentations, typical lesions, and alternative diagnoses that have been ruled out. By demonstrating the presence of OCB, we may provide proof for the disease’s immunological and inflammatory nature without waiting for the spread to occur. Thus, OCB is a well-established biomarker with clinical relevance for MS diagnosis [143,144,145,146].
2.4.1. IgG Index
The immunoglobulin (Ig) G index is defined as the ratio of IgG’s CSF/serum quotient to albumin’s CSF/serum quotient. The albumin quotient, defined as albumin in CSF divided by albumin in serum, is used to assess blood-CSF barrier failure in MS [147]. The IgG index is used to quantify intrathecal immunoglobulin synthesis. An IgG index result greater than 0.7 implies an elevated intrathecal B cell response and, consequently, MS’s existence. Around 70% of people with MS have a high IgG index. As a result, this biomarker’s sensitivity is lower than that of the OCB [148,149].
2.4.2. Antinuclear antibodies
Antinuclear antibodies (ANA) are tissue-independent autoantibodies directed against components of the cell nucleus that are quantified in the serum [150]. A continuously high titer indicates collagenous SLE [151].
2.4.3. Anti-MOG antibodies
MOG is a myelin protein that is mostly located on the surface of myelin sheaths and oligodendrocyte membranes, and it could be a target for the autoimmune response in demyelinating disorders [152,153]. Therefore, anti-MOG antibodies, contrary to common perception, are only beneficial for differential diagnosis, not for MS diagnosis or prognosis. Using cutting-edge detection technologies, anti-MOG antibodies were found in a subset of pediatric patients with acute disseminated encephalomyelitis (ADEM), patients with clinical symptoms of NMOSD, and patients with bilateral optic neuritis in particular (cell-based approaches) [154].
2.4.4. Anti-aquaporin-4 antibodies
Aquaporin-4 (AQP-4) is a water channel protein that is expressed by astrocytes in the CNS and is necessary for brain water homeostasis [155,156]. Antibodies to this protein are found in around 75% of patients with neuromyelitis optica spectrum disorder (NMOSD), but not in MS patients. Anti-aquaporin-4 antibodies are thus appropriate for high-specificity biomarkers. It is the first molecular biomarker to be clinically proven for differentiating between distinct CNS inflammatory demyelinating disorders. Antibodies against aquaporin-4 are frequently seen in the serum of patients suspected of having NMOSD [157].
This entry is adapted from the peer-reviewed paper 10.3390/molecules27113516
This entry is offline, you can click here to edit this entry!