representing almost 90% of the world market. While the acceptance of biosimilars in the US is catching up with that in the EU, the cost benefits remain elusive due to the high development barrier and complex distribution system involved, mainly in the US. In the EU, the cost of biosimilars has already dropped 70% or more, and interchangeability is a routine in some European jurisdictions, unlike in the US, where a separate regulatory approval is required. This paper projects significant changes coming in the US and EU’s biosimilars approval requirements that will impact the approval procedures in the rest of the world, leading to dramatic changes in the cost of biosimilars to patients. This perspective is based on the author’s first-hand experience to secure FDA approvals of biosimilars and an extensive analysis of the rationality of testing to demonstrate biosimilarity. Multiple citizen petitions by the author and meetings with the FDA may have prompted the recent announcement by the FDA to award a $5 million research grant to scientists to develop novel testing models to establish biosimilarity, including modifying the interchangeability protocols. Soon, demonstration of biosimilarity will not require animal testing and, in most cases, clinical efficacy testing; over time, the clinical pharmacology testing will be reduced as the regulatory agencies develop more confidence in the safety and efficacy of biosimilars. Biosimilars have come of age; now it is the turn of the developers to grow up, and one way to show this is to challenge the current regulatory guidelines but only on scientific grounds to seek more concessions, for which both FDA and EMA are ready.
- biosimilars
- FDA
- EMA
- Clinical Efficacy
- Analytical Assessment
- Interchangeability
- Animal Toxicology
- Biosimilarity
1. Introduction
2. Target Molecules
| Biological Products | |||
|---|---|---|---|
| Abatacept | Abciximab | Aflibercept | Alemtuzumab |
| Alirocumab | Atezolizumab | Avelumab | Basiliximab |
| Bedinvetman (V) | Belimumab | Benralizumab | Bevacizumab |
| Bezlotoxumab | Blinatumomab | Blood factors | Brentuximab vedotin |
| Brodalumab | Brolucizumab | Burosumab | Canakinumab |
| Caplacizumab | Cemiplimab | Certolizumab pegol | Cetuximab |
| Crizanlizumab | Daclizumab | Daratumumab | Darbepoetin alfa |
| Denosumab | Dinutuximab | Dupilumab | Durvalumab |
| Eculizumab | Elotuzumab | Emapalumab | Emicizumab |
| Erenumab | Etanercept | Evolocumab | Follitropin alfa |
| Fremanezumab | Frunevetmab (V) | Galcanezumab | Gemtuzumab ozogamicin |
| Golimumab | Guselkumab | Ibalizumab | Idarucizumab |
| Inotuzumab ozogamicin | Insulin detemir | Insulin lispro | Interferons |
| Ipilimumab | Isatuximab | Ixekizumab | Lanadelumab |
| Lokivetab (V) | Mepolizumab | ogamulizumab | Moxetumomab pasudodox |
| Muromonab-CD3 | Natalizumab | Necitumumab | Nivolumab |
| Obiltoxaximab | Obinutuzumab | Ocrelizumab | Ofatumumab |
| Olaratumab | Omalizumab | Palivizumab | Panitumumab |
| Pembrolizumab | Pertuzumab | Polatuzumab vedotin | Ramucirumab |
| Ranibizumab | Ravulizumab | Raxibacumab | Reslizumab |
| Rilonacept | Risankizumab | Romosozumab | Sacituzumab govitecan-hziy |
| Sarilumab | Secukinumab | Selumetinib | Siltuximab |
| Teprotumumab-trbw | Tildrakizumab | Tocilizumab | Urofollitropin |
| Ustekinumab | Vedolizumab | ||
3. Patent Litigation
4. Biosimilar Adoption
4.1. The US Scene
-
“Anti-competitive practices, such as making false or misleading statements comparing biological reference products and biosimilars, may be slowing progress and hampering the uptake of these important therapies”, quoted from an FDA and Federal Trade Commission (FTC) joint statement [22] made in February 2020.
-
The FDA also agreed to “take appropriate steps to address companies making false or misleading communications about biologics, including biosimilars and interchangeable products, which will help deter anti-competitive behavior in the biologics market and lead to the use of all available biological products”, according to the statement. In a news release dated 20 July 2021, the FDA stated that Amgen is making false claims regarding its Neulasta medicine being more effective in its new delivery system Onpro, citing this joint statement [23].
-
President Biden signed an executive order titled “Promoting Competition in the American Economy” [24], which directs the Federal Trade Commission to issue rules to prevent “unfair anticompetitive conduct or agreements in the prescription drug industries, such as agreements to delay the market entry of generic drugs or biosimilars”. The order also directs the FDA to address several issues affecting biosimilars, including: (1) “improving and clarifying interchangeability standards for biological products”; (2) “supporting biosimilar product adoption by providing effective educational materials and communications to improve understanding of biosimilar and interchangeable products among healthcare providers, patients, and caregivers”; and (3) “facilitating the development (by sponsors) and approval (acceptance) of biosimilar and interchangeable products among healthcare providers, patients, and caregivers”. Status: enacted.
-
A new law, the “Advancing Education on Biosimilars Act” [25], now calls for the government to provide educational materials to healthcare providers, patients, and the general public to increase awareness, knowledge, and confidence in the safety and efficacy of approved biosimilars. Status: enacted.
-
The “Star Rating for Biosimilars Act” [26], recently presented, adds a qualification system to Medicare plans. Status: introduced.
-
The “Bolstering Innovative Options to Save Immediately on Medicines” (BIOSIM) Act [27] intends to lower biologic drug prices by temporarily increasing reimbursement to ASP plus 8% (from ASP plus 6% previously) for providers that employ a biosimilar that is less expensive than the reference product. Status: introduced.
-
The “Preserve Access to Affordable Generics and Biosimilars Act” [28] changes the Federal Trade Commission Act to presumptively render anticompetitive “pay-for-delay” (also known as “reverse-payment”) settlement agreements that prohibit or delay the introduction of generic pharmaceuticals or biosimilars. Status: enacted.
4.2. The European Scene
5. Regulatory Pathway
Both the FDA and the EMA are now ready to accept applications with a substantially reduced quantity of data; this change will significantly impact the cost of developing biosimilars, as described in a recent McKinsey report [8].
5.1. The US Scene
There are over 100 biosimilar programs enrolled with the FDA [33]. To expedite the approval process, the FDA has taken several significant steps.
- The FDA has created two new guidelines, the extension of the Q&A presentations [34] and the third revised draft guidance [35] titled “New and Revised Draft Q&As on Biosimilar Development and the BPCI Act”. The details refer to fulfilling pediatric assessment or PREA requirements, post-approval filing, and the assertion that the 351(k) cannot have a different route or dosage form. However, the strength issue was delayed, adding new indications and orphan exclusivity. The FDA also updated The Purple Book FAQ section [36].
- FDA has also published new fact sheets [37] to provide additional educational materials on biosimilar and interchangeable products and the biosimilar regulatory review and approval process. The BPCIA states [38] that the “Secretary may determine, in the Secretary’s discretion, that an element described in clause (i) (I) [the biosimilar testing] is unnecessary in an application submitted under this subsection”. The FDA has subtly implemented this change in its new biosimilar guidance [39].
- The BPCIA text [38] states that “an application submitted under this subsection shall include information demonstrating that the biological product is biosimilar to a reference product based upon data derived from analytical studies, animal studies, and clinical studies”. The new education material includes the phrase “in addition to analytical studies, other studies that may be needed”, not shall be, as stated in the BPCIA.
- Animal studies are now described as unnecessary for providing toxicology or pharmacology information about a biosimilar.
- Clinical pharmacology studies show that the proposed biosimilar passes through the body the same way as the reference product and has the same effects. This could include an immunogenicity test to see how the biosimilar affects a patient’s immune system.
- Additional clinical studies can sometimes be conducted after other studies to address any remaining uncertainty about whether the proposed biosimilar has clinically meaningful differences from the reference product.
The historic pyramid of the FDA is now replaced with a crescent [40] (Figure 2).
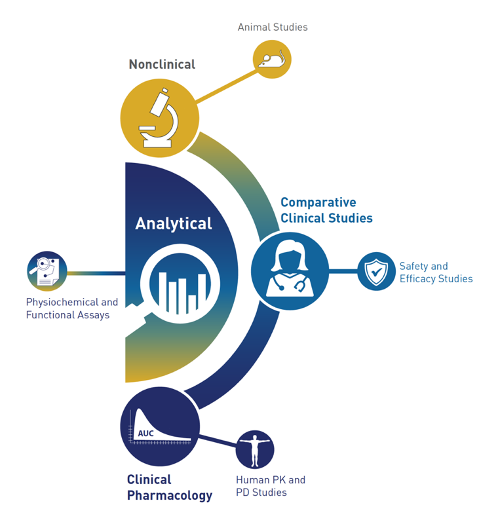
Figure 2. The new FDA rollout of the development crescent for biosimilars (FDA: Level 1 Biosimilar and Interchangeable Products Foundational Concepts (https://www.fda.gov/drugs/biosimilars/curriculum-materials-health-care-degree-programs-biosimilars; accessed on 4 April 2022).
5.2. The European Scene
The EU was the first region to develop a robust regulatory framework for the authorization of biosimilars. In 2001, much of the EU’s directive-based legislation concerning the regulation of medicines was codified as Directive 2001/83/EC [41]. The European Medicines Agency (EMA) has developed a regulatory scheme for biosimilar authorization through a relatively transparent process. The EMA has issued concept papers and draft guidance and held public scientific workshops. It has issued guidelines that describe general principles and provided an overarching framework for the authorization of biosimilars. The EMA’s Committee for Medicinal Products for Human Use (CHMP) has also issued product class-specific guidance that sets out product requirements in greater detail. For example, the CHMP has issued guidelines for recombinant erythropoietin, granulocyte-colony stimulating factor, recombinant human soluble insulin, low-molecular-weight heparins, somatropin, and recombinant interferon alfa [42]. EMA has announced that they intend not to issue more specific biosimilar guidelines but instead prefer to give tailored advice on a case-by-case basis [43].
It is anticipated that the EMA will add more guidelines, particularly for monoclonal antibodies, that will significantly reduce the regulatory barrier and the cost and time taken to reach the market. The FDA has stayed away from creating product-specific guidelines and discouraged the USP from creating any monographs for biological drugs to prevent the originators from including specifications that might be protected intellectual property.
In 2022, the CHMP initiated a new program to engage additional stakeholders during the discussions relating to the evaluation of the development of products; it is anticipated that more changes to the regulatory control of biosimilars will result from this effort [42]. In addition, the EPAR program of CHMP is an excellent source of information and learning for biosimilar developers; 84 dossiers are available [44]. The FDA also posts details of its approval of biosimilar products. However, a biosimilar developer may object to the posting, in which case the details can only be secured under the Freedom of Information Act [45].
6. Analytical Assessment
The analytical assessment includes testing physicochemical and functional attributes to establish a claim of biosimilarity. How closely a biosimilar candidate should match the reference product will remain questionable since a reference product is approved based on whatever quality attributes it presents; a biosimilar candidate, on the other hand, must match these quality attributes, even if the reference product’s attributes are not the most desirable. An earlier FDA guideline, “Statistical Approaches to Evaluate Analytical Similarity” [46], recommended a rigorous statistical approach for establishing similarity that turned out to be overkill, and the guidance was withdrawn [47] and replaced with a new guideline [48] in response to the author’s citizen petition [49]. The new guideline changed the terminology from “analytical testing” to “analytical assessment”, meaning an overall evaluation rather than specific test results. For example, this eliminated the controversial tier 1 assessment of quality attributes. In addition, this required setting up arbitrary equivalence criteria such as 1.5 × SD of the reference product to define the 90% confidence limit of the biosimilar candidate, with no justification for the factor of 1.5 used. Instead, the new guideline suggests using a range approach that is more practical and scientifically sound. However, as all biosimilar products approved by the FDA followed the earlier guideline, there is a lot of analytical testing that would be avoidable in the future. For example, companies have submitted different number of studies for adalimumab—25 by Pfizer and 71 by Boehringer—to achieve the same goal [43].
The EMA provides more comprehensive guidance divided into immunogenicity testing, quality issues, clinical and non-clinical testing, pharmacokinetic modeling, and guidance on changing the manufacturing process of recombinant drugs [50]. In addition, the product-specific guidelines of the EMA are of great value for biosimilar developers [51].
Most regulatory guidelines suggest that a biosimilar candidate’s quality target product profile (QTPP) should be based on the data collected on the chosen reference product, including publicly available information and data obtained from the extensive characterization of the reference medicinal product [52]. The QTPPs are well defined, and there may not be any need to establish their relative importance and assign a criticality factor to plan the testing, as these are now well-established. However, as suggested below, the developers should challenge the merits of testing an attribute. Quality attributes fall into two categories, product- or process-related.
6.1. Product-Related Attributes
Product-related attributes (not to be confused with the drug product that is the finished form) relate to the production of proteins by cells that can make exact copies of the protein [40] (Figure 3). Still, after the protein is made, other variations (e.g., add-ons and changes) may occur, such as adding sugar molecules or modifying certain amino acids. The expression system determines the product-related attributes with as little manipulation as possible. The QTPP profile must match the reference product and undergo the well-established testing required. Tests of the biosimilar must be conducted side-by-side with tests of the reference product to remove any test method variability, as the test methods need not be validated (Figure 3).
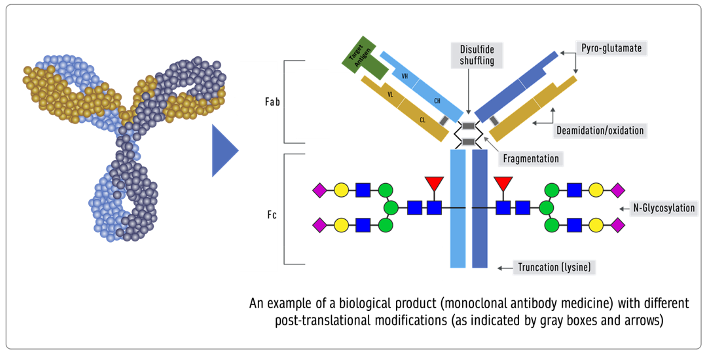
Figure 3. Inherent variations in biological products (FDA: https://www.fda.gov/drugs/biosimilars/curriculum-materials-health-care-degree-programs-biosimilars; accessed on 4 April 2022) [40].
- Peptide mapping (LC-MS), peptide mass fingerprinting (MALDI-MS), MALDI TOF, and MS amino acid sequencing are all examples of primary structure sequencing.
- Higher-order structures can be confirmed using thermodynamic DSC, NMR, SPR, ELISA, fluorescence, far and near UV CD, DSC, NMR, SPR, and ELISA. While process-related testing is straightforward and well-established, testing product-related attributes can be improved by testing the UV and fluorescence spectra under various stress conditions, temperature surfactants, electrolytes, and pH [53]. Newer and more sensitive methods are always needed.
- Cell-based assays, SPR, and ELISA, to test receptor binding.
- Forced degradation: degradation is forced to match intramolecular bond strength as a structural similarity measure.
6.2. Process-Related Attributes
The process-related quality attributes are dependent on the manufacturing process used; thus, they are made part of the release specification to assure compliance. Establishing the acceptance criteria for these quality attributes can be achieved based on legacy values, as is considered to be standard practice for injectable products or the criteria established by testing the reference product.
Ideally, a process-related attribute should be made part of the release specification. The release limits can be derived from legacy values (previously established and known) or by testing the reference product. The European and British Pharmacopoeias [54] have developed monographs of several key biological products defining quality attributes to establish release specifications. The USP has stated that it will not develop monographs for a biologic unless there is stakeholder consensus supporting its creation, including the support of the FDA [55]. The FDA has discouraged the USP from creating biologics monographs to ensure that innovator biologics makers do not use the monograph process to block biosimilar competition by incorporating patented characteristics of their product that are not relevant to safety, purity, or potency, thereby further impacting competition [56].
However, despite the different opinions on using a monograph to develop a biosimilar product, many legacy attributes, the quality attributes that come from historical and experience-based variability, are widely accepted as norms.
- Protein content. Biological products label potency of 100 IU/mL for insulin in vials. Based on shared experience, the protein content cannot always be the same due to filling variability, concentration testing variability, and many other unpredictable factors. For this reason, most products are allowed an acceptable practical range of variability of ±5% [57]. However, this quality attribute is controversial, as the first FDA guideline required this attribute to be tested for equivalence. The 95% CI of the biosimilar product cannot go beyond 1.5*SD of the reference product in an equivalence test. This range was established entirely arbitrarily. If the SD of the reference product turns out to be small, all batches of the biosimilar product will fail despite being within the release specification of ±5%. This means that a biosimilar product might be acceptable for patients but not for approval by the FDA. This situation arose when the first biosimilar EP2006 required the testing of 50 lots to match the equivalence criteria of Amgen’s Neupogen, despite all lots meeting the release specifications [58]. We can use this as an example to remove the comparative testing of the protein content from side-by-side testing. However, if a biosimilar product has a higher variability, this must be confirmed with the variability in the reference product lots.
- Post-translation modifications, aggregates, and isomers should be tested in a range model, wherein 90% of the values of the biosimilar lots should fall within 3 × SD of the reference product to establish analytical similarity and the specification should include a range of no more than 3 × SD of the reference product.
- Bioassay limits are calculated as specified in the statistical analysis of biological assays and test results. They are typically expressed as an acceptable range for the estimated potency (e.g., 80–125 percent of the stated potency) and an acceptable range for the confidence limits of the estimated potency (e.g., 64–156 percent of the stated potency) [59].
- Impurities in biological products, also known as residuals, are of much greater importance than in chemical drugs. Impurities can be either process- or product-related. Process-related impurities are derived from the manufacturing process—for example, cell culture, downstream, or cell substrates. In contrast, product-related impurities are non-active molecular variants of the biologic and are formed during expression, manufacture, or storage. Understanding these impurities is essential to developing control strategies to reduce or remove them from the final product. The impurities caused by the upstream process may include cell culture reagents, antifoams, growth modifiers (insulin), antibiotics, protein a, solubilizers, residual solvents, chelating agents, extractable extracts, and leachable. The downstream-derived impurities may include detergent, protein a, process additives, chromatographic resins, extractable, and leachable. Cell-derived impurities include host cell DNA and host cell proteins. Product-related impurities include truncated forms such as fragments; modified forms such as disulfide, oxidation, deamidation, and glycosylation; and aggregates including multimers and subvisible particles. When present in a substantial quantity, these impurities may reduce the product’s potency and, worse, induce immunogenic responses or alter the product’s pharmacokinetics. While process-related impurities can be readily isolated, product-related impurities are often difficult to separate because of their close structural similarity to the active molecules. As a result, a biosimilar product must not have any unmatched impurity. There is also no analytical method or biological test that can ensure the safety of an unmatched impurity since any testing of immunogenicity in an animal species may not match the immune response in humans. In some cases, an unmatched impurity may be acceptable if the same regulatory agency has approved an identical structure or there is sufficient published proof of its safety. Since matched impurities can reduce efficacy if they are not as efficacious, a variation of 3% is generally allowed as a legacy attribute. Additionally, the 3% variation must not include more than 1% of any single impurity. However, these acceptance criteria can also be established by profiling the reference product.
- Particle size (subvisible), residual DNA, fill volume, and sterility standards are well defined in several official compendia, and these should be acceptable.
- Physical properties. If the formulation is the same, then the formulation’s physical properties, such as surfactants, osmolality, and pH, should fall within three standard deviations of that of the reference product. However, when the formulation is different, the release specifications will be based on testing multiple lots of biosimilar products. The BPCIA allows a biosimilar product to have a different formulation; however, using the same formulation as the reference product reduces the risk of higher immunogenicity, especially if the inactive component(s) are used in another biological product and have the same route of administration. This is in contrast to the WHO’s suggestion that “relevant differences in formulation (for example, use of excipients in the biosimilar that are not widely used in medicinal products)” can be tested using animal models [60], despite experience gained from the incidence of immunological reactions induced by erythropoietin formulations that used a different formulation [61]. No animal testing can establish the safety of inactive components when used in a biological drug formulation.
Since analytical similarity assessment is the core of biosimilar product evaluation, most regulatory audits pertain to these details after filing the registration application. They often result in multiple complete response letters (CRLs) that delay approval. This includes data integrity and CFR 21 Part 11 compliance, proof of test method suitability (suitable or validated), and blinding issues. Therefore, outsourcing the analytical assessment may be more cost-effective and time-effective. First, this realization regarding the analytical assessment audits came after multiple products were filed, and several qualified CDMOs can now fulfill this role.
7. Nonclinical Pharmacology
Testing in animals is an old routine used for new drugs [62] to avoid serious toxicity to humans. However, biological drugs may not always show a pharmacologic response in animal species; thus, the toxicity is an extension of the pharmacological response for biological drugs. The primary mechanism of action of biological drugs involves receptor binding. Suppose an animal species does not carry these receptors. Then, a pharmacological or toxicological response is not expected, unlike in chemical drugs, where both can be caused by multiple mechanisms and interactions with body tissues [63].
Another reason animal toxicology data are less relevant is how the testing is conducted. Generally, animal testing protocols require administering a higher dose to induce a toxic response; however, within this dose range, the responses are not expected to be linear, making it impossible to differentiate between compared products that are supposed to be the same. Animal testing is extensively conducted for biosimilars despite this knowledge and expertise, evidenced by the recent FDA and EMA filings. As an example, MVASI (bevacizumab) [64] reported five non-clinical investigations; the product Trazimera (trastuzumab) [65] was tested in mice to justify the receptor binding discrepancies, even though trastuzumab does not recognize the neu receptor; and another similar product submitted by Herzuma included many more animal studies [66], which the FDA did not review as irrelevant. Similar submissions were made for Renflexis and Inflectra, two biosimilars of infliximab [67, 68], and the etanercept biosimilars Eticovo and Erelzi [69, 70]. The highest number of animal studies, 15, was recorded for the epoetin biosimilar Retacrit [71]. The non-clinical development of the two pegfilgrastim biosimilars, Udenyca and Fulphilia [72, 73], is notable for the different animal models; Udenyca reported a toxicity investigation in Cynomolgus monkeys, whereas Fulphilia reported a toxicity study in rats. The trend shown here for the US biosimilar filings extends to Europe, where, despite the availability of waivers from animal testing, the developers continued the testing [74].
Another controversial issue in animal studies is the use of non-human primates, which are the only species that may have relevant receptors; it is frequently recommended to conduct PK studies in a small number of animals, especially for monoclonal antibodies, as a measure of their molecular structure rather than toxicity. According to the WHO [60], “based on regulatory experience gained to date in marketing authorization applications for biosimilars, the need for additional in vivo animal studies would be expected to represent a rare scenario”. However, the guidelines in India take a very different view, stating, “Regarding the animal models to be used, the applicant should provide the scientific justification for the choice of animal model(s) based on the data available in scientific literature. However, if the pharmacologically relevant animal species are not available and appropriately justified, toxicity studies need to be undertaken either in rodent or non-rodent species” [75]. This requirement was put in place because India requires at least one animal toxicology study, and no studies are allowed on monkeys for religious reasons.
Stronger support for waiving animal pharmacology or toxicology testing comes from the recent advice by regulatory bodies suggesting that it is unnecessary to test new biological therapies in animals, even if an animal species can show a response unless there is carcinogenicity potential [76].
Generally, it is now believed that testing new drugs in animal species may lead to misguidance if the safety is based on animal testing, resulting in serious threats [77]. In addition, humanized or genetically modified animal species are generally considered to be less sensitive in demonstrating differences in the tested products [78]. With this evolving background, the testing of biosimilars seems redundant, as stated in the European Union legislation on the protection of animals used for scientific purposes [79] and the FDA/CDER advocacy to use new approach methodologies (NAMs) [80] in place of animal testing.
Human and animal cells, organoids, organs-on-chips, and in silico modeling are alternatives to animal testing models, enabling us to create better and more predictive scientific methods. In addition, to reflect changes in animal protection legislation, nonclinical in vivo testing has been substituted by in vitro assays in the previous 10 years [81]. These measures can help to reduce the use of animals. They also align with the EMA’s Regulatory Science Strategy for 2025, aiming to create a more adaptive regulatory framework that promotes human and veterinary health [82].
Animal toxicological studies can be misleading if they rationalize discrepancies in impurities, posttranslational modifications, or antibody responses, since an animal model can justify these differences. For example, animal data were submitted in biosimilar applications [66] to substantiate such variability, but the FDA refused to accept the animal data.
More than 100 products have been approved by the EMA and FDA, and none of them have failed animal toxicological testing because they cannot, being least sensitive in detecting any difference between a biosimilar candidate and its reference product. These observations and conclusions are widely accepted as scientifically sound arguments [83, 84], but among sponsors, there is always fear that study results will be rejected eventually. This would cause a delay in market access at a high cost, and therefore sponsors like to stay on the safe side by overpowering their studies.
8. Clinical Pharmacology
Clinical pharmacology comparisons comprise the most relevant testing to support the biosimilarity of a biosimilar candidate. When a novel drug is developed, PK/PD testing is carried out on many volunteers to understand the diversity of disposition in terms of gender, age, and genetic distribution. However, such a population is not needed for establishing biosimilarity; the purpose of clinical pharmacology studies for a new drug is to characterize its profile. In the case of biosimilars, it compares the profile. A smaller number of subjects can be enrolled in these studies by narrowing down the acceptance criteria that will be acceptable since agencies also recommend that there be no unnecessary exposure for humans [85]. This suggestion reduces the risk of study failure without compromising the purpose of these studies—to compare how the body sees the molecule and how the molecule sees the body.
Besides reducing the size of the study, as described above, this study model can combine antidrug antibody measurements in a parallel design that should be presented to regulatory agencies earlier in meetings with them [87].
The FDA Biosimilar Action Plan [86] also recommends employing in silico methodologies to compare biosimilars, including immunogenicity assessments. Since immunogenicity is entirely structure-dependent, better analytical assessment techniques give greater confidence in reducing or eliminating antidrug antibody testing. In addition, impurities and aggregates induce extrinsic immunogenicity, which may be easily measured and compared to a reference product as part of the analytical evaluation.
The immunogenicity of biological products is caused by the activation of B cells, which generate T cells to express antibodies. However, anti-drug antibodies can be harmful to healthy subjects in future studies. As a result, the FDA is researching new methods for determining immunogenic potential using tiny fragments of DNA-like molecules called aptamers to test proteins and establish their exact structures to avoid the exorbitant costs of forecasting which particular portions of such proteins will stimulate antibody production [87].
Finally, if the immunogenicity profile differs but cannot impact the disposition profile, the differences will be meaningless, as the FDA has acknowledged in its new guidance on insulins [88, 89].
Following the idea that humans should not be subjected to unnecessary testing, the FDA has agreed to allow non-US reference products as long as they are approved using “essentially” the same dossier [90] and if an analytical bridging study is also conducted. However, many developers have instead chosen to conduct three-way studies using US and non-US reference standards; such studies are unnecessary.
PK/PD studies are essentially bioequivalence testing using the same statistical limits of 80–125% bioequivalence, a guideline that arose in the era of generic chemical drugs. Intravenously administered drugs were exempt from bioequivalence testing because, by definition, they are 100% bioequivalent. However, in the case of the evaluation of biosimilars, this testing is intended as an additional assurance of structural similarity, which relates to how the body sees the molecule and how the molecule sees the body. I anticipate that our analytical assessment will become more convincing over time, which will enable us to waive these studies.
9. Clinical Efficacy and Safety
“If there is residual uncertainty about biosimilarity after conducting structural analyses, functional assays, animal testing, human PK and PD studies, and the clinical immunogenicity assessment, the sponsor should then consider what additional clinical data may be needed to address that uncertainty (section VII.D.3) adequately”, according to the BPCIA. However, having additional clinical data does not necessarily imply a clinical efficacy investigation; it might include in silico pharmacokinetic research, as indicated by the FDA in its Biosimilar Action Plan [86]. EMA stated that “generally, clinical data aim to address slight differences shown at previous steps and to confirm the comparable clinical performance of the biosimilar and the reference product”. Clinical data cannot be used to justify substantial differences in quality attributes [91]. Therefore, the first argument relates to identifying “slight differences” or, as the FDA labels it, “residual uncertainty”. Are we not able to ascertain these differences and uncertainties? If so, then clinical trials are irrelevant. If there are no differences, why test, and if there are differences, why not reject them?
Clinical safety and efficacy studies add substantial cost and time to the approval of biosimilars. However, this argument will have little weight if these studies were able to add additional value over and above the rest of the testing. Thus far, no biosimilar products have been rejected based on clinical efficacy and safety testing if they passed the rest of the testing. This means either the products were biosimilar or the testing was too insensitive to detect any difference [92, 93]. In both cases, this testing becomes irrelevant. This concept of real-time testing is now also questioned by the FDA, which stated that clinical efficacy testing is “broken” [94] and that new digital technologies and real-world evidence (RWE) are required, as outlined in the 21st Century Cure Act [95].
The European Medicines Agency (EMA) has begun work on a pilot clinical trial program aiming to advise how to decrease or eliminate clinical testing in biosimilar development [96]. Comparative clinical trials are increasingly seen as sloppy techniques for assessing biological agent similarity. As a result, the testing of biosimilars in patients is more of a checkmark than a meaningful indication.
Biosimilars “may be approved based on PK and PD biomarker data without a comparative clinical study with efficacy endpoint(s)”, according to FDA guidance [97]. The use of PK and PD biomarker data in healthy participants or patients enables shorter and less expensive clinical investigations and provides more sensitive testing than clinical efficacy with endpoint(s), as demonstrated with filgrastim [98]. The FDA acknowledged this and granted approval for filgrastim-aafi, filgrastim-sndz, pegfilgrastim-jmdb, pegfilgrastim-cbqv, and epoetin alfa-epbx based solely on PD evaluation. Furthermore, the FDA identified the features of PD biomarkers in its advice to assist sponsors in using PD biomarkers as part of biosimilar development programs [96].
Another reason why the clinical efficacy testing of biosimilars can be fallacious is due to the testing models used: equivalence or non-inferiority. In the equivalence testing mode, we first determine the M1 or total efficacy value of the reference product—a highly variable but available parameter; second, we select an acceptable range of difference, the M2, based on a clinical judgment that usually cannot be definitive—at best, it is an arbitrary choice. As a result, since both products are expected to be identical, equivalency studies are least likely to fail. On the other hand, non-inferiority testing is contraindicated because a biosimilar product showing a higher efficacy may also have more safety issues.
Many drugs, including anticancer drugs, require the homogeneity of the study population, which is unlikely. Patients are inevitably exposed to multiple drugs and treatment modalities; additionally, anticancer drugs have a low efficacy rate, further reducing the statistical probability of identifying any difference. Oncology or other terminal illness treatment efficacy studies face specific hurdles, such as enrolling a comparable group of naive patients. Such investigations further fail due to the brief lifespans of patients, which can disrupt the study design.
Another argument against clinical efficacy testing is the extrapolation of indications allowed for the biosimilar product. If there are any doubts about the safety or efficacy, then they should be tested in all indications, not just one selected by the developer, even where the modes of action are the same. A good example is conducting a psoriasis study for adalimumab approval instead of testing in psoriatic arthritis.
10. Interchangeability
Interchangeability is a legally defined path in the US biosimilar guidance. While it does not fall under the responsibility of the EMA in Europe, the practice of interchangeability means that the individual member states are left to decide their policies regarding switching and substitution. In addition, while the FDA has singular guidance, in the EU, there are many different frameworks and available advice, making this a highly complex issue [99]. In several European countries, switching and substitution are forced, and for years this practice has resulted in no untoward effects. For example, in Denmark and Norway, the interchangeability is automatic without consultation with prescribers or patients.
In the US, an interchangeable status can be secured for an approved biosimilar after switching and alternating studies to assure that the response from a biosimilar product will be the same as that of the reference product every time. In July 2021, the FDA approved the first interchangeable biosimilar in the U.S. for Viatris’ Semglee (insulin glargine-yfgn), referencing the long-acting insulin, Lantus. The approval was significant for a multitude of reasons: Not only is Semglee the first interchangeable biosimilar but the first biosimilar in diabetes care and the first biosimilar that is primarily dispensed at retail pharmacies; therefore, it is billed under the pharmacy benefit.
A new federal executive order requires the FDA to clarify the assignment of interchangeability status. The first clarification needed is that a biosimilar product is expected to have “no clinically meaningful difference” from the reference product. How this determination differs from interchangeability is not discussed by the FDA. Second, since many biological products are given a single dose, how would one test switching and alternating protocols? The anticipated guideline changes in the FDA are likely to allow developers to conduct simpler testing to qualify for the interchangeability status that awards them exclusivity for automatic substitution as the first to secure this status [100].
A common misconception is that interchangeable biosimilars must meet higher standards for approval than non-interchangeable biosimilars. However, all biosimilars—whether interchangeable or not—undergo rigorous testing.
11. Development Perspective
Making biosimilars accessible means reducing their cost of development, which is currently at around USD 100–200 million, keeping small and medium-size companies out of play and leaving most current biosimilars in the hands of big pharma. How this cost breaks down is an interesting subject; for example, a recent study [101] reported a median (IQR) estimated cost of USD 20.8 (USD 13.8–35.3) million and a median (IQR) treatment duration of 52 (28–68) weeks; when switching and alternating, the cost was USD 27.6 (USD 18.0–36.7) with a median (IQR) treatment duration of 55 (46–78) weeks. The trial duration included the period needed to establish the effectiveness and the extensions during which patients were switched between products. For oncology product trials, which typically continue indefinitely, the trial duration was defined as the period from the date of the reported trial start to the date when the FDA accepted the data. For the two hematopoietic products for which the FDA did not require testing in patients, the cost was for a median (IQR) treatment duration of 15 (14–15) weeks, with a median (IQR) estimated cost of USD 1.9 (USD 1.6–1.9) million. Interestingly, the cost of similar studies for new molecular entities was similar to or even lower than that of the comparative testing since a much larger population of patients is required to establish the statistical significance of findings when the two arms are supposed to be providing an equivalent response. At the same time, the clinical pharmacology studies recruited about 100 participants, with more than 500 patients on average included in the clinical efficacy testing [92].
According to the data reported in ClinicalTrials.gov, 667 clinical studies involving biosimilars were reported [93], 598 were listed as interventional, and 68 were listed as observational. The number of studies conducted was 891 due to the multiple sites involved. The number of studies that reported their testing phase included early phase 1, 4; phase 1, 189; phase 2, 281; phase 3, 163; phase 4, 15; and phase not applicable, 9. There seems to be some discord in defining the study phase; in some, no early phase or phase 2 study is required, and even some listed as phase 3 can more appropriately be called a comparative efficacy study. Assuming the costs of studies as suggested above are not out of the ballpark, these studies must have cost over USD 10 billion, which is not a large number for big pharma. However, to see smaller companies entering the field of biosimilars, reducing the cost of clinical testing (except clinical pharmacology) will be a significant motivation.
The current estimates of the cost of a new biosimilar product coming to market at USD 100–200 million are overestimated since these are based on the cost factors associated with big pharma operations. One of the larger cost elements is the depreciation of the CAPEX, which can quickly run into several hundred million dollars. This number is based on the experience of the author. Additionally, the cost adds up if the submission takes longer and FDA audits and approval are delayed for various reasons, as mentioned above. For example, holding multiple FDA or EU meetings will lead to a longer submission time. Each meeting takes a 4–5 month toll; now that the approval pathway is clear, intelligent regulatory planning could quickly reduce the filing to 18 months. Other delays may come from patent litigation and whether the developer chooses to submit the filing to the originator company.
Choosing the product for development is another dilemma for many since development costs are identical regardless of the potential market. It is no surprise that the market leaders such as adalimumab, with current sales of over USD 18 billion, are the most popular biosimilars. However, the situation with adalimumab will change starting in 2025 when approved biosimilars that are held back due to litigation will hit the market. The total market of adalimumab is then expected to decrease by 50%. Table 2 lists the projected sales in the year 2025 and current approvals in EU and US [101].
Table 2. The projected market of biologicals in the year 2025 as impacted by the entry of biosimilars and the development factor.
|
No |
Product (Brand) Company |
Global (Billion USD) Market, 2025 1 [101] |
Current Approved US/EU Biosimilars 2 [9,10] |
Development Factor 3 |
|
1. |
Erythropoietin (Epoetin) Amgen |
18 |
1/3 |
1 (anemia) |
|
2. |
Pembrolizumab (Keytruda), Merck |
16 |
0/0 |
5 (oncology) |
|
3. |
Nivolumab (Opdivo), BMS |
14 |
0/0 |
5 (oncology) |
|
4. |
Adalimumab (Humira) AbbVie |
11 |
7/10 |
2 (TNF) |
|
5. |
Etanercept (Enbrel), Amgen |
8 |
2/3 |
2 (TNF) |
|
6. |
Infliximab (Remicade), Janssen |
8 |
4/4 |
2 (TNF) |
|
7. |
Ustekinumab (Stelara), Janssen |
7.5 |
0/0 |
2 (TNF) |
|
8. |
Bevacizumab (Avastin) Roche |
7 |
3/9 |
4 (oncology) |
|
9. |
Ocrelizumab (Ocrevis) |
7 |
0/0 |
3 (MS) |
|
10. |
Pertuzumab (Perjeta) Roche |
7 |
0/0 |
5 (oncology) |
|
11. |
Secukinumab (Cosentyx) |
6 |
0/0 |
2 (TNF) |
|
12. |
Aflibercept (Eyelea), Regeneron |
4 |
0/0 |
2 (AMD) |
|
13. |
Darbepoetin alfa (Aranesp) Amgen |
4 |
0/0 |
1 (anemia) |
|
14. |
Peg-filgrastim (Neulasta), Amgen |
4 |
4/7 |
1 (neutropenia) |
|
15. |
Ranibizumab(Lucentis) Novartis |
4 |
1/1 |
2 (AMD) |
|
16. |
Trastuzumab (Herceptin), Genentech |
4 |
5/6 |
4 (oncology) |
|
17. |
Rituximab (Rituxan) Biogen |
3 |
3/5 |
4 (oncology) |
|
18. |
Cetuximab (Erbitux): (Lilly/Merck) |
1 |
0/0 |
5 (oncology) |
|
19. |
Eculizumab (Soliris) Alexion |
1 |
0/0 |
3 (hemoglobinuria) |
1: Market data from open source; 2: Biosimilar approved in US and EU based on data as of April 2022 posted by the FDA and EMA; 3: “Development Factor” is a term coined to project the time and cost to market, 1 = lowest; 5 = highest, assessed by the author.
I am also presenting a parameter, “development factor”, to indicate the cost and time factor to take a biosimilar to the market. The primary consideration is the phase 3 study; in some cases, such as the TNF products, an efficacy study can be a smaller psoriasis study, but the oncology drugs will remain at a high development cost, at least for now. The lower development factor also comes for products with PD or clinical markers that are easier to monitor.
Since the cost to take a product to market depends on building a sound regulatory plan, one comes across difficulties in complying with the different global authorities, which seem to have divergent requirements; this prevents many companies from going global with their biosimilars.
Monoclonal antibodies comprise the majority of biological products. It is now well established that the manufacturing cost of these antibodies is USD 95–200 per gram, regardless of the type of antibody involved [102]. For oncology antibodies, the dosing is generally 150–800 mg [103]. As an example, Rituxan (rituximab) DS is priced at USD 10,000 per gram [104]. This should encourage developers, as they will have a substantial margin even at a 70–80% price reduction.
12. Recommendations
Biosimilars have come of age; now is the developer’s turn to make them accessible. A few recommendations taken from the experience of the last 17 years of the life of biosimilars and a longer engagement by the author in their development teach us that:
- Since 60% of all new drugs are biologics, there will be a long list of eligible biosimilars for the future.
- More than 100 biological products have expired patents and expired exclusivity waiting for biosimilar candidacy.
- Veterinary biological products are additional choices for biosimilars that have been neglected.
- It will take a price drop of 70% or more across all biological products to make biosimilars accessible to all. However, many countries have already reached this stage.
- The COGs of all antibodies are between USD 95 and 200 per gram, and they are priced at 100×; despite the price drop, there will still be high profit margins.
- The adoption of biosimilars will require taking stakeholders into confidence, particularly prescribers and patients.
- Countries where forced switching and alternating are doing just as well despite restrictions.
- Global markets will require approval from the EU and US. Both agencies offer fee-free advice. Design studies are acceptable in both the EU and US. US protocols will likely be acceptable to the EMA, but not the other way round.
- Regulatory guidelines are neither binding on the agencies nor the developers. Therefore, we need to question them, challenge them, and create a rational development plan that does not originate from the agencies.
- Biosimilars and interchangeable product guidelines will undergo substantial revision, reducing the burden of testing and replacing it with advance testing tools.
- An analytical assessment is most pivotal to approval; we need to adopt newer technologies and plans, not redundant testing. We can reduce testing by limiting product-related attributes. We can outsource analytical assessments to avoid delays in regulatory approval.
- Do not offer to conduct any animal testing; it is not the role of regulatory agencies to tell companies what not to do.
- Design creative clinical pharmacology protocols to reduce the size of studies and secure all data from one study.
- Do not offer to conduct clinical efficacy testing and challenge the suggestion made by the regulatory agencies to identify the “residual uncertainty”.
- If a clinical efficacy test must be conducted, choose an indication where markers are better defined to reduce the study size, such as using psoriasis to test adalimumab.
The best evidence to support my perspective that changes are coming in the regulatory guidelines came in March 2022, when the FDA announced a grant of USD 5 million for a variety of project types, including analytical methodology (including bioassay) development, in silico tools, real-world evidence, pharmacology studies, and ancillary studies in parallel to planned or ongoing clinical trials and combinations of these project types. In some cases, funding of a novel pharmacokinetic/pharmacodynamic study may be considered. The FDA is particularly interested in projects that efficiently and convincingly achieve intended objectives. Therefore, novel, efficient, and convincing strategies to validate such tools and standards are welcome. A novel method or tool without validation or a feasible approach to validation will not be acceptable [105].
Now that biosimilars have come of age, it is time for developers to grow up [106].
Funding: This research received no external funding.
Institutional Review Board Statement: This research received no external funding.
Informed Consent Statement: Not Applicable
Data Availability Statement: Not Applicable
Conflicts of Interest: The author declares no conflict of interest.
References
- Omnitrope Drug Approval Package. The US. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021426s000TOC.cfm (accessed on 23 March 2022).
- Zarxio Drug Approval Package. US. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=125553 (accessed on 23 March 2022).
- FDA Approves Cyltezo, the First Interchangeable Biosimilar to Humira. Second Interchangeable Biosimilar Product Approved by FDA. US. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-cyltezo-first-interchangeable-biosimilar-humira (accessed on 23 March 2022).
- How the U.S. Compares to Europe on Biosimilar Approvals and Products In the Pipeline (Updated). JDSUPRA. Available online: https://www.jdsupra.com/legalnews/how-the-u-s-compares-to-europe-on-6251301/ (accessed on 23 March 2022).
- The Impact of Biosimilar Competition in Europe December 2021. IQVIA. Available online: https://www.iqvia.com/-/media/iqvia/pdfs/library/white-papers/the-impact-of-biosimilar-competition-in-europe-2021.pdf (accessed on 23 March 2022).
- Kansteiner, F. JP Morgan 2022: Jan 11, 2022. Amgen Dials up Biosimilar Ambitions, with at Least $4B in Expected Sales by 2030. Available online: https://www.fiercepharma.com/pharma/jpm-2022-amgen-aims-to-double-biosimilar-sales-by-2030 (accessed on 23 March 2022).
- Medicare Part B Drug Average Sales Price, Center for Medicare and Medicaid Services. Available online: https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Part-B-Drugs/McrPartBDrugAvgSalesPrice (accessed on 23 March 2022).
- Chen, Y.; Monnard, A.; da Silva, J.S. An Inflection Point for Biosimilars. McKinsey & Co. 7 June 2021. Available online: https://www.mckinsey.com/industries/life-sciences/our-insights/an-inflection-point-for-biosimilars (accessed on 23 March 2022).
- Biosimilars Approved in Europe (Updated 28 January 2022), Generic and Biosimilar Initiative. Available online: https://gabionline.net/biosimilars/general/biosimilars-approved-in-europe (accessed on 23 March 2022).
- Biosimilar Product Information. Available online: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information (accessed on 4 April 2022).
- Amgen Biosimilar Trend Report. Amgen Biosimilars. Available online: https://www.amgenbiosimilars.com/-/media/Themes/Amgen/amgenbiosimilars-com/Amgenbiosimilars-com/pdf/USA-CBU-80961_Amgen-Biosimilars-Trend-Report.pdf (accessed on 23 March 2022).
- Generic Enoxaparin Questions and Answers. Available online: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/generic-enoxaparin-questions-and-answers (accessed on 23 March 2022).
- FDA Statement: Insulin Gains New Pathway to Increased Competition. Available online: https://www.fda.gov/news-events/press-announcements/insulin-gains-new-pathway-increased-competition (accessed on 23 March 2022).
- Biosimilars to Continue Rapid Growth over the Next Decade. IQVIA. Available online: https://www.iqvia.com/blogs/2021/12/biosimilars-to-continue-rapid-growth-over-the-next-decade (accessed on 23 March 2022).
- Drug Patent Watch. Available online: https://www.drugpatentwatch.com (accessed on 23 March 2022).
- Biologics Market Dynamics: Setting the Stage for Biosimilars. Available online: https://www.ftc.gov/system/files/documents/public_events/1568297/aitken_-_biologics_market_dynamics_setting_the_stage_for_biosimilars_slides.pdf (accessed on 23 March 2022).
- Biologics Market--Growth, Trends, COVID-19 Impact, and Forecasts. (2022–2027). Mordor Intelligence. Available online: https://www.mordorintelligence.com/industry-reports/biologics-market#:~:text=The%20biologics%20market%20was%20valued,forecast%20period%2C%202021%2D2026 (accessed on 23 March 2022).
- BPCIA Litigation. Big Molecule Watch. Available online: https://www.bigmoleculewatch.com/bpcia-patent-litigations/(accessed on 23 March 2022).
- Kracov, D.; Marsh, D.; Tabas, M.; Ho, T. FDA Seeks to Deepen Engagement with USPTO. 27 September 2021. Available online: https://www.arnoldporter.com/en/perspectives/publications/2021/09/fda-seeks-to-deepen-engagement-with-uspto (accessed on 23 March 2022).
- Biosimilars Year in Review 2021, Fish & Richardson. Available online: https://www.fr.com/biosimilars-2021-year-in-review/(accessed on 23 March 2022).
- Association for Accessible Medicines. 2021 U.S. Generic and Biosimilar Medicines Savings Report. Retrieved from Association for Accessible Medicines: Generics & Biosimilars. October 2021. Available online: https://accessiblemeds.org/sites/default/files/2021-10/AAM-2021-US-Generic-Biosimilar-Medicines-Savings- Report-web.pdf (accessed on 4 April 2022).
- FDA Press Announcement: FDA and FTC Announce New Efforts to Further Deter Anticompetitive Business Practices. Available online: https://www.fda.gov/news-events/press-announcements/fda-and-ftc-announce-new-efforts-further-deter-anti-competitive-business-practices-support (accessed on 23 March 2022).
- FDA Notifies Amgen of Branding its Product Neulasta. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-notifies-amgen-misbranding-its-biological-product-neulasta-due-false-or-misleading-promotional (accessed on 4 April 2022).
- Presidential Order on Promoting Competition in the American Economy. Available online: https://www.whitehouse.gov/briefing-room/presidential-actions/2021/07/09/executive-order-on-promoting-competition-in-the-american-economy/ (accessed on 23 March 2022).
- US Senate: Advancing Education on Biosimilars Act. Available online: https://www.cassidy.senate.gov/imo/media/doc/Advancing Education on Biosimilars Act.pdf (accessed on 23 March 2022).
- US Congress. Star Rating for Biosimilars Act. Available online: https://www.congress.gov/bill/117th-congress/house-bill/2855?q=%7B%22search%22%3A%5B%22Star+Rating+for+Biosimilars+Act%22%2C%22Star%22%2C%22Rating%22%2C%22for%22%2C%22Biosimilars%22%2C%22Act%22%5D%7D&s=1&r=1 (accessed on 4 April 2022).
- US Congress. H.R.2815—BIOSIM Act. Available online: https://www.congress.gov/bill/117th-congress/house-bill/2815/text?r=1&s=1 (accessed on 23 March 2022).
- US Congress. Preserve Access to Af3 Fordable Generics and Biosimilars Act. Available online: https://www.congress.gov/117/bills/s1428/BILLS-117s1428rs.pdf (accessed on 23 March 2022).
- Rémuzat, C.; Kapuśniak, A.; Caban, A.; Ionescu, D.; Radière, G.; Mendoza, C.; Toumi, M. Supply-side and demand-side policies for biosimilars: An overview in 10 European member states. Mark. Access Health Policy 2017, 5, 1307315.
- Moorkens, E.; Vulto, A.G.; Huys, I.; Dylst, P.; Godman, B.; Keuerleber, S.; Claus, B.; Dimitrova, M.; Petrova, G.; Sović-Brkičić, L.; et al. Policies for biosimilar uptake in Europe: An overview. PLoS ONE 2017, 12, e0190147.
- Love, B. US Plays catch-up with Europe over Biosimilar Patents, Financial Times. 16 June 2021. Available online: https://www.ft.com/content/3f7ca3f4-8256-4570-a6a3-b255e185f162 (accessed on 23 March 2022).
- Moorkens, E.; Vulto, A.G.; Huys, I. An overview of patents on therapeutic monoclonal antibodies in Europe: Are they a hurdle to biosimilar market entry? MAbs 2020, 12, 1743517.
- FDA-TRACK: Center for Drug Evaluation & Research—Pre-Approval Safety Review—Biosimilars Dashboard. Available online: https://www.fda.gov/about-fda/fda-track-agency-wide-program-performance/fda-track-center-drug-evaluation-research-pre-approval-safety-review-biosimilars-dashboard (accessed on 23 March 2022).
- Questions and Answers on Biosimilar Development and the BPCI Act Guidance for Industry. September 2021. Available online: https://www.fda.gov/media/119258/download (accessed on 23 March 2022).
- New and Revised Draft Q&As on Biosimilar Development and the BPCI Act (Revision 3) Guidance for Industry. September 2021. Available online: https://www.fda.gov/media/119278/download (accessed on 23 March 2022).
- Purple Book. Available online: https://purplebooksearch.fda.gov/faqs#5 (accessed on 4 April 2022).
- Healthcare Providers Materials. Available online: https://www.fda.gov/drugs/biosimilars/health-care-provider-materials?utm_medium=email&utm_source=govdelivery#fact (accessed on 23 March 2022).
- Title VII—Improving Access to Innovative Medical Therapies Subtitle A—Biologics Price Competition and Innovation. US Congress. Available online: https://www.fda.gov/media/78946/download (accessed on 23 March 2022).
- Biological Regulatory Review and Approval. FDA. Available online: https://www.fda.gov/media/151061/download (accessed on 4 April 2022).
- Level 1 Biosimilar and Interchangeable Products Foundational Concepts. FDA. Available online: https://www.fda.gov/drugs/biosimilars/curriculum-materials-health-care-degree-programs-biosimilars (accessed on 23 March 2022).
- Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use Official Journal L–311, 28/11/2004, pp. 67–128. EMA. Available online: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/directive-2001/83/ec-european-parliament-council-6-november-2001-community-code-relating-medicinal-products-human-use_en.pdf (accessed on 23 March 2022).
- European Medicines Agency. Human Regulatory. Biosimilars. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/multidisciplinary/multidisciplinary-biosimilar#-product-specific-biosimilar-guidelines-section (accessed on 23 March 2022).
- Wolff-Holz, E.; Tiitso, K.; Vleminckx, C.; Weise, M. Evolution of the EU Biosimilar Framework: Past and Future. BioDrugs 2019, 33, 621–634. https://doi.org/10.1007/s40259-019-00377-y.
- European Medicines Agency. EPAR Biosimilars. Available online: https://www.ema.europa.eu/en/search/search/field_ema_web_categories%253Aname_field/Human/ema_group_types/ema_medicine/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar?search_api_views_fulltext=biosimilar%20monographs (accessed on 23 March 2022).
- Freedom of Information Act. Available online: https://www.fda.gov/regulatory-information/freedom-information (accessed on 23 March 2022).
- Guideline on Statistical Approaches to Evaluate Analytical Similarity Guidance for Industry. 2017. Available online: https://www.pbwt.com/content/uploads/2018/06/UCM576786.pdf (accessed on 23 March 2022).
- FDA Withdraws Draft Guidance for Industry: Statistical Approaches to Evaluate Analytical Similarity. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-withdraws-draft-guidance-industry-statistical-approaches-evaluate-analytical-similarity (accessed on 23 March 2022).
- Development of Therapeutic Protein Biosimilars: Comparative Analytical Assessment and Other Quality-Related Considerations Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein-biosimilars-comparative-analytical-assessment-and-other-quality (accessed on 23 March 2022).
- Forbes Magazine. One Man’s Mission to Fix the FDA’s Biosimilar Problem. Available online: https://www.forbes.com/sites/nicolefisher/2018/07/25/one-mans-mission-to-fix-the-fdas-biosimilar-problem/?sh=1843e1723808 (accessed on 23 March 2022).
- Niazi, S. Analysis of FDA-Licensed Biosimilars: Time for a Paradigm Shift. AJMC, Center for Biosimilars. Available online: https://www.centerforbiosimilars.com/view/analysis-of-fda-licensed-biosimilars-time-for-a-paradigm-shift (accessed on 23 March 2022).
- European Medicines Agency Biotechnology Products. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-0.pdf (accessed on 23 March 2022).
- Vandekerckhove, K.; Seidl, A.; Gutka, H.; Kumar, M.; Gratzl, G.; Keire, D.; Coffey, T.; Kuehne, H. Rational Selection, Criticality Assessment, and Tiering of Quality Attributes and Test Methods for Analytical Similarity Evaluation of Biosimilars. AAPS J. 2018, 20, 68. https://doi.org/10.1208/s12248-018-0230-9.
- Niazi, S. Methods for Comparing a Structure of a First Biomolecule and a Second Biomolecule. US Patent Application 20140356968, 2014. Available online: https://tinyurl.com/h58tdjnr Accessed (accessed on 23 March 2022).
- European Directorate for the Quality of Medicines & HealthCare. Biotherapeutics Monographs. Available online: https://www.edqm.eu/sites/default/files/medias/bio_tab_portfolio_january_2022.pdf (accessed on 4 April 2022).
- Statement on Monographs for Biologics. US Pharmacopoeia. Available online: https://www.usp.org/news/statement-on-monographs-for-biologics (accessed on 23 March 2022).
- FDA-USP Clash over Biologics Monographs. Available online: https://www.raps.org/news-and-articles/news-articles/2019/6/fda-usp-clash-over-biologics-monographs (accessed on 4 April 2022).
- Goyal, P.; Pai, H.V.; Kodali, P.; Vats, B.; Vajpai, N.; Annegowda, S.; Mane, K.; Mohan, S.; Saxena, S.; Veerabhadraia, A.B.; et al. Physicochemical and functional characterization of MYL-1501D, a proposed biosimilar to insulin glargine. PLoS ONE 2021, 16, e0253168.
- Oncology Briefing Document. 15 January 2015. Available online: https://patentdocs.typepad.com/files/briefing-document.pdf (accessed on 23 March 2022).
- European Directorate for the Quality of Medicines & HealthCare. Synthetic Peptides and rDNA Products. Available online: https://www.edqm.eu/sites/default/files/guide_ph_eur_synthetic_peptides_and_rdna_proteins_2018.pdf (accessed on 23 March 2022).
- WHO Guidelines on Evaluation of Biosimilars. World Health Organization. Available online: https://cdn.who.int/media/docs/default-source/biologicals/who-guidelines-on-evaluation-of-biosimilars---4-nov-2021.pdf?sfvrsn=f17799ae_5 (accessed on 23 March 2022).
- McKoy, J.M.; Stonecash, R.E.; Cournoyer, D.; Rossert, J.; Nissenson, A.R.; Raisch, D.W.; Casadevall, N.; Bennett, C.L. Epoetin-associated pure red cell aplasia: Past, present, and future considerations. Transfusion 2008, 48, 1754–1762. https://doi.org/10.1111/j.1537-2995.2008.01749.x.
- Hajar R. Animal testing and medicine. Heart Views. 2011, 12, 42.
- Niazi, S. Debate over Animal Toxicology Studies. AJMC. Center for Biosimilars. Available online: https://www.centerforbiosimilars.com/view/opinion-the-debate-over-animal-toxicology-studies (accessed on 23 March 2022).
- Drug Approval Package: Mvasi (bevacizumab-awwb). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761028Orig1s000TOC.cfm (accessed on 23 March 2022).
- Drug approval package: Trazimera (trastuzumab-qyyp). Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=761081 (accessed on 4 April 2022).
- Drug Approval Package: Herzuma (tratuzumabn-pkrb). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761091Orig1s000TOC.cfm (accessed on 4 April 2022).
- Drug Approval Package: Renflexis (Infliximab-abda). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761054Orig1s000TOC.cfm (accessed on 4 April 2022).
- Drug Approval Package. Inflectra (infliximab-dyyb) for injection. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/125544Orig1s000TOC.cfm (accessed on 4 April 2022).
- Drug Approval Package: Eticovo. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761066Orig1s000TOC.cfm (accessed on 4 April 2022).
- Drug Approval Package: Erelzi (etanercept-szzs). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761042Orig1_toc.cfm (accessed on 4 April 2022).
- Drug Approval Package: Retacrit (epoetin alfa-epbx). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/125545Orig1s000TOC.cfm (accessed on 4 April 2022).
- Drug Approval Package: Udenyca. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761039Orig1s000TOC.cfm (accessed on 4 April 2022).
- Drug Approval Package: Fulphila. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761075Orig1s000TOC.cfm (accessed on 23 March 2022).
- Pipalava, P.; Patel, R.; Mehta, M.; Dahiya, M.; Singh, I.; Jose, V. An update on the animal studies conducted for biosimilar approvals—Regulatory requirement vs. actual scenario. Toxicol. Pharmacol. 2019, 107, 104415. https://doi.org/10.1016/j.yrtph.2019.104415.
- CDSCI India. Guidelines on Similar Biologics. Available online: https://birac.nic.in/webcontent/Guidelines_on_Similar_Biologics_06_10_2017.pdf (accessed on 23 March 2022).
- van Aerts, L.A.; De Smet, K.; Reichmann, G.; van der Laan, J.W.; Schneider, C.K. Biosimilars entering the clinic without animal studies, a paradigm shift in the European Union. MAbs 2014, 6, 1155–1162.
- Van Norman G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach? JACC Basic Transl. Sci. 2019, 4, 845–854. https://doi.org/10.1016/j.jacbts.2019.10.008.
- Bailey, J. Chapter 19: Genetic Modification of Animals: Scientific and Ethical Issues. In Animal Experimentation: Working towards a Paradigm Change; Brill: Leiden, The Netherlands. 2019; pp. 443–479, ISBN: 9789004391192. https://doi.org/10.1163/9789004391192_020.
- European Medicines Agency. EMA Implements New Measures to Minimise Testing During Medicines Development. European Medicines Agency. Available online: https://www.ema.europa.eu/en/news/ema-implements-new-measures-minimise-animal-testing-during-medicines-development (accessed on 23 March 2022).
- Advancing Alternate Methods at FDA. Available online: https://www.fda.gov/science-research/about-science-research-fda/advancing-alternative-methods-fda (accessed on 23 March 2022).
- European Medicines Agency. Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the Protection of Animals Used for Scientific Purposes Text with EEA Relevance. European Medicines Agency. Directive 2010/63/EU. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32010L0063 (accessed on 23 March 2022).
- European Medicines Agency. Regulatory Science Strategy. European Medicines Agency. Available online: https://www.ema.europa.eu/en/about-us/how-we-work/regulatory-science-strategy#regulatory-science-strategy-to-2025-section (accessed on 23 March 2022).
- Schiestl, M.; Ranganna, G.; Watson, K.; Jung, B.; Roth, K.; Capsius, B.; Trieb, M.; Bias, P.; Maréchal-Jamil, J.; The Path Towards a Tailored Clinical Biosimilar Development. BioDrugs 2020, 34, 297–306.
- Evaluating Inclusion and Exclusion Criteria in Clinical Trials. Available online: https://www.fda.gov/media/134754/download (accessed on 23 March 2022).
- Niazi, S. Testimony to the US. Available online: https://www.regulations.gov/document/FDA-2019-P-1236-0003 (accessed on 4 April 2022).
- Biosimilars Action Plan. FDA. Available online: https://www.fda.gov/media/114574/download (accessed on 4 April 2022).
- Immunogenicity of Protein-Based Therapeutics. FDA. June 2020. Available online: https://www.fda.gov/vaccines-blood-biologics/biologics-research-projects/immunogenicity-protein-based-therapeutics (accessed on 23 March 2022).
- Keown, A. FDA Allows Waiver of Clinical Trials for Insulin Biosimilars as Recommended in Niazi Citizen Petition. BioSpace. Published 3 December 2019. Available online: https://www.biospace.com/article/releases/fda-allows-waiver-of-clinical-trials-for-insulin-biosimilars-as-recommended-in-niazi-citizen-petition/ (accessed on 23 March 2022).
- Clinical Immunogenicity Considerations for Biosimilars and Interchangeable Insulin Products. FDA. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-immunogenicity-considerations-biosimilar-and-interchangeable-insulin-products (accessed on 23 March 2022).
- Guidance: Clinical Pharmacology Data to Demonstrate Biosimilarity to a Reference Product. FDA. 2016. Available online: https://www.fda.gov/media/88622/download (accessed on 23 March 2022).
- European Medicines Agency. Tailored Scientific Advice for Biosimilars Development. Available online: https://www.ema.europa.eu/en/documents/report/tailored-scientific-advice-biosimilar-development-report-experience-pilot-2017-2020_en.pdf (accessed on 23 March 2022).
- Moore, T.J.; Mouslim, M.C.; Blunt, J.L.; Alexander, G.C.; Shermock, K.M. Assessment of Availability, Clinical Testing, and US Review of Biosimilar Biologic Products. JAMA Intern. Med. 2021, 181, 52–60. https://doi.org/10.1001/jamainternmed.2020.3997.
- Biosimilars Clinical Testing Registered. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=biosimilar&cntry=&state=&city=&dist= (accessed on 23 March 2022).
- Brennan, J. FDA’s Woodcock Says the Clinical Trial System Is Broken. Regulatory Affairs Professionals Society. Available online: https://www.raps.org/regulatory-focus%E2%84%A2/news-articles/2017/9/fda-s-woodcock-the-clinical-trials-system-is-broken (accessed on 23 March 2022).
- 21st Century Cures Act. Available online: https://www.fda.gov/regulatory-information/selected-amendments-fdc-act/21st-century-cures-act (accessed on 23 March 2022).
- Li, J.; Florian, J.; Campbell, E.; Schrieber, S.J.; Bai, J.P.; Weaver, J.L.; Hyland, P.L.; Thway, T.M.; Matta, M.K.; Lankapalli, R.H.; et al. Advancing Biosimilar Development Using Pharmacodynamic Biomarkers in Clinical Pharmacology Studies. Pharmacol. Ther. 2020, 107, 40–42. https://doi.org/10.1002/cpt.1653.
- FDA Guidance: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. FDA. 2015. Available online: https://www.fda.gov/media/82647/download (accessed on 23 March 2022).
- Li, L.; Ma, L.; Schrieber, S.J.; Rahman, N.A.; Deisseroth, A.; Farrell, A.T.; Wang, Y.; Sinha, V.; Marathe, A. Quantitative relationship between AUEC of absolute neutrophil count and duration of severe neutropenia for G-CSF in breast cancer patients. Pharmacol. Ther. 2018, 104, 742–748.
- Barbier, L.; Mbuaki, A.; Simoens, S.; Declerck, P.; Vulto, A.G.; Huys, I. Regulatory Information and Guidance on Biosimilars and Their Use Across Europe: A Call for Strengthened One Voice Messaging. Available online: https://www.frontiersin.org/articles/10.3389/fmed.2022.820755/full (accessed on 23 March 2022).
- Glintborg, B.; Loft, A.G.; Omerovic, E.; Hendricks, O.; Linauskas, A.; Espesen, J.; Danebod, K.; Jensen, D.V.; Nordin, H.; Dalgaard, E.B.; et al. To switch or not to switch: Results of a nationwide guideline of mandatory switching from originator to biosimilar etanercept. One-year treatment outcomes in 2061 patients with inflammatory arthritis from the DANBIO registry. Ann Rheum Dis. 2019, 78, 192–200. https://doi.org/10.1136/annrheumdis-2018-213474.
- European Pharmaceutical Review. Top 10 Drugs by Annual Revenue in 2025. Available online: https://www.europeanpharmaceuticalreview.com/article/102539/top-10-drugs-by-annual-revenue-in-2025/ (accessed on 4 April 2022).
- Call for Consultant on Monoclonal Antibodies for Infectious Diseases. Available online: https://www.who.int/news-room/articles-detail/call-for-consultant-on-monoclonal-antibodies-for-infectious-diseases (accessed on 23 March 2022).
- Hendrikx, J.J.M.A.; Haanen, J.B.A.G.; Voest, E.E.; Schellens, J.H.M.; Huitema, A.D.R.; Beijnen, J.H. Fixed Dosing of Monoclonal Antibodies in Oncology. Oncologist 2017, 22, 1212–1221. https://doi.org/10.1634/theoncologist.2017-0167.
- Price of Rituxan. Available online: https://www.webmd.com/rx/drug-prices/rituxan (accessed on 23 March 2022).
- Biosimilar User Fee Act (BsUFA) Research Grant (U01) Clinical Trials Optional. Available online: https://grants.nih.gov/grants/guide/rfa-files/RFA-FD-22-026.html (accessed on 14 April 2022).
- Niazi, S.K., Biosimilars: A futuristic fast-to-market advice to developers, Expert Opinion on Biological Therapy, 22:2, 149-155. Available on line: https://www.tandfonline.com/doi/citedby/10.1080/14712598.2022.2020241?scroll=top&needAccess=true . (accessed 14 April, 2022). DOI: 1080/14712598.2022.2020241.
This entry is adapted from the peer-reviewed paper 10.3390/biologics2020009