Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Periodontitis is one of the most common oral diseases resulting in gingival inflammation and tooth loss. Growing evidence indicates that it results from dysbiosis of the oral microbiome, which interferes with the host immune system, leading to bone destruction. Immune cells activate periodontal ligament cells to express the receptor activator of nuclear factor kappa-B (NF-κB) ligand (RANKL) and promote osteoclast activity. Osteocytes have active roles in periodontitis progression in the bone matrix. Local proteins are involved in bone regeneration through functional immunological plasticity.
- periodontitis
- osteoimmunology
- bone regeneration
- osteoblast
- osteocyte
- osteoclast
1. Introduction
The human oral cavity contains many microbial species constituting commensal bacteria, with interactions between the host and oral microbiota defining oral cavity health. The oral microbiota is a distinct and diversified ecosystem of microbial organisms that each microorganism interacts with, metabolically and physically. These complex interactions culminate in the creation of biofilm communities in which physiochemical gradients establish diverse habitats for microorganisms requiring varying metabolic activities [1]. The microbiota structure in healthy supragingival plaque resembles the “hedgehog-like” network, resulting from the radially spatio-chemical gradient [2]. In the structure depicting the plaque microbiota described by Welch et al. [2], early colonizers, such as Actinomyces spp. and Streptococcus spp., adhere to the tooth surface via interaction of non-specific and specific binding adhesins on their cell surfaces and salivary proteins within the pellicle [3]. Consequently, Corynebacterium spp. attaches to the early colonizers and radially grows exteriorly to form a long, annular structure. Haemophilus, Aggregatibacter, and Neisseriaceae attach to the tip of the annulus owing to their metabolic activity requiring the abundance of both oxygen and nutrients. Metabolic products from oxidative species at the periphery create anoxic circumstances in the core of the biofilm, where anoxic capnophilic species, such as Capnocytophaga and Fusobacterium, prefer to grow [2]. Unlike many infectious diseases, periodontitis results from oral microbiota dysbiosis [4][5][6]. The etiology of periodontal diseases is due to periodontal-pathogenic bacteria. When the sophisticated interactions of the oral microbiota are disturbed by the keystone bacterium, Porphyromonas gingivalis, the polymicrobial dysbiosis transpires [7][8]. This disturbance in plaque microbiota is a significant etiology of gingival inflammation causing the onset of periodontitis [8][9][10]. Oral microbiota dysbiosis interferes with the host immune system, resulting in inflammatory conditions, in turn leading to cellular interactions of immune and bone cells and subsequent bone destruction [11][12]. The cellular interactions and molecules related to the communication between immune and bone cells have gained popularity in recent years, providing more insights into periodontitis.
Osteoimmunology focuses on the cellular and molecular mechanisms behind inflammatory bone resorption, which destroys alveolar bone [13]. Recent studies in osteoimmunology focused on understanding the pathogenesis and development of therapies for inflammatory bone diseases, such as rheumatoid arthritis and periodontitis [14].
2. Periodontitis: Osteoimmunology
2.1. RANKL and OPG
Bone is a dynamic tissue resulting from calcium and collagen homeostasis through balanced osteoclast and osteoblast function. Osteoclasts derived from monocytes/macrophages are multinucleated cells that resorb bone matrix. Osteoblasts control osteoclastogenesis via the production of macrophage colony-stimulating factor (M-CSF) and RANKL, an essential cytokine in the activation of osteoclasts [14][15][16]. Alveolar loss results from dominant osteoclast activity. This activity depends on the interaction of three proteins composing the RANK/RANKL/OPG axis [17][18]. Transmembrane receptor activator of nuclear factor-κB (RANK) receptor is expressed in progenitor and mature osteoclasts with RANK binding to its ligand, RANKL, determining osteoclast activity. RANKL is a cytokine in the TNF family [13] that activates the RANK receptor [19]. However, osteoprotegerin (OPG) is a soluble decoy receptor for RANKL, impeding the RANK/RANKL binding and inhibiting osteoclastogenesis [20]. The RANKL/OPG ratio increases at periodontitis-affected sites, emphasizing the importance of the equilibrium between the molecules in this axis, especially RANKL and OPG levels [21][22].
Membrane-bound RANKL is responsible for almost all primary functions; however, soluble RANKL has a minor role in physiological bone homeostasis in mice [23]. Depriving soluble RANKL in the mouse model did not influence the level of bone destruction, which emphasizes the role of membrane-bound RANKL in the pathology of osteoporosis and periodontitis [15][23][24]. Accordingly, cells expressing membrane-bound RANKL are either near the bone surface or in contact with it [14]. exFoxp3Th17 cells exhibit the highest mRNA and protein levels of RANKL amongst T cell subsets [15]. Moreover, Th17 and exFoxp3Th17 cells produce IL-17 to activate mesenchymal cells, such as osteoblasts and PDL cells, to express RANKL on their membrane and produce proinflammatory cytokines to stimulate osteoclastic differentiation following alveolar bone loss [15][25]. Therefore, cells expressing membrane-bound RANKL derived near the periodontitis-affected bone surface lead to periodontitis. In addition, Th17 cells and exFoxp3Th17 T cell subsets likely contribute to mesenchymal cell stimulation via IL-17 to express RANKL, followed by stimulation of osteoclast functions.
Reduced OPG levels result in an elevated RANKL/OPG ratio, with a human study demonstrating significantly decreased OPG mRNA expression and OPG immunostaining in periodontitis lesions compared with the healthy periodontium [26]. In addition, proteases derived from oral bacteria and osteoclasts cleave OPG and stimulate osteoclast function in vitro [27][28]. This emphasizes the important roles of each protein in controlling osteoclast function in the axis, which is the critical activity affecting periodontitis.
2.2. Osteocytes Are Not Just Quiescent Resident Cells
Periodontitis is a multidimensional disease causing the tissue and teeth-supporting bone to disorganize. The pathophysiological process of periodontal disease has primarily focused on bone loss, particularly osteoclastogenesis and osteoblastogenesis. Recent studies show that osteocytes are not quiescent and contribute to physiological and pathological events in periodontitis [29].
Osteocytes are derived from an osteoblast cell lineage differentiated from mesenchymal stem cells (MSCs) in the bone marrow and account for most of the bone cells. They have an extended lifetime of up to 25 years compared with a half-life of 150 days for osteoblasts [30]. When osteoblasts cease to secrete extracellular matrix (ECM), they either undergo apoptosis or differentiate into osteocytes [31][32]. Communication between osteocytes and other cells can occur via dendrites reaching the surface of the bone (the lacunocanalicular system) [32]. This connection establishes crosstalk between osteoclasts, osteoblasts, bone lining cells, and bone marrow cells with the osteocytes [30][32][33]. Osteoclasts affect bone metabolism via osteocyte connectors. Osteoclast-derived leukemia inhibitory factor reduces sclerostin expression in osteocytes and subsequently promotes osteoblastic bone formation [34][35].
2.3. Osteocytes Induce Osteoclastogenesis via M-CSF (CSF-1) and RANKL
Osteoclasts are multinucleated giant cells originating from monocyte/macrophage cells and are accountable for bone resorption. Characteristics of periodontitis progression include alveolar bone loss resulting from osteoclast differentiation or osteoclastogenesis [36]. Osteoclastogenesis occurs via the production of colony-stimulating factor-1, also known as M-CSF, a factor required for osteoclast differentiation [37][38]. Continuous production of M-CSF in osteocytes enhances osteoclastogenesis [39]. Osteocyte-derived M-CSF protects against excessive Nox4-derived ROS generation and retains bone remodeling [40].
RANKL is a membrane-associated cytokine required for osteoclastogenesis synthesized by osteoblasts and osteocytes [37]. Osteoclastogenesis driven by RANKL is critical for inflammatory bone resorption, and its expression increases in periodontitis [41][42]. RANKL is synthesized by osteoblasts and interacts with the RANK receptor on the cell membrane of pre-osteoclasts to initiate conversion into active osteoclasts. OPG is the naturally present RANKL decoy receptor and is generated by various osteoblast lineages [43]. OPG competes with and reduces RANKL binding to RANK, inhibiting the activation of osteoclasts [44].
Osteocytes produce RANKL during bone remodeling in periodontitis (Figure 1). The concept of osteoblasts being the primary cellular source of RANKL has shifted toward osteocytes, which serve as the primary source of RANKL in bone remodeling instead of osteoblasts [45][46]. Gram-negative bacteria-derived lipopolysaccharide interacts with toll-like receptors on the osteocyte cell surface to stimulate the mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) 1/2 signaling pathway resulting in the activation of transcription factors that upregulate IL-6 expression [47]. IL-6 then activates Janus kinase through gp130, phosphorylating signaling molecules, and activator of transcription (STAT), leading to the translocation of STAT into the nucleus, enhancing RANKL expression in osteocytes [48][49]. Apart from IL-6, TNF-α is a typical inflammatory cytokine in periodontitis that directly stimulates osteocytes to produce RANKL and induce osteoclastogenesis or promote sclerostin expression in osteocytes leading to increased osteoclastogenesis [39]. Nevertheless, TNF-α does not induce osteocytes to produce M-CSF [50].
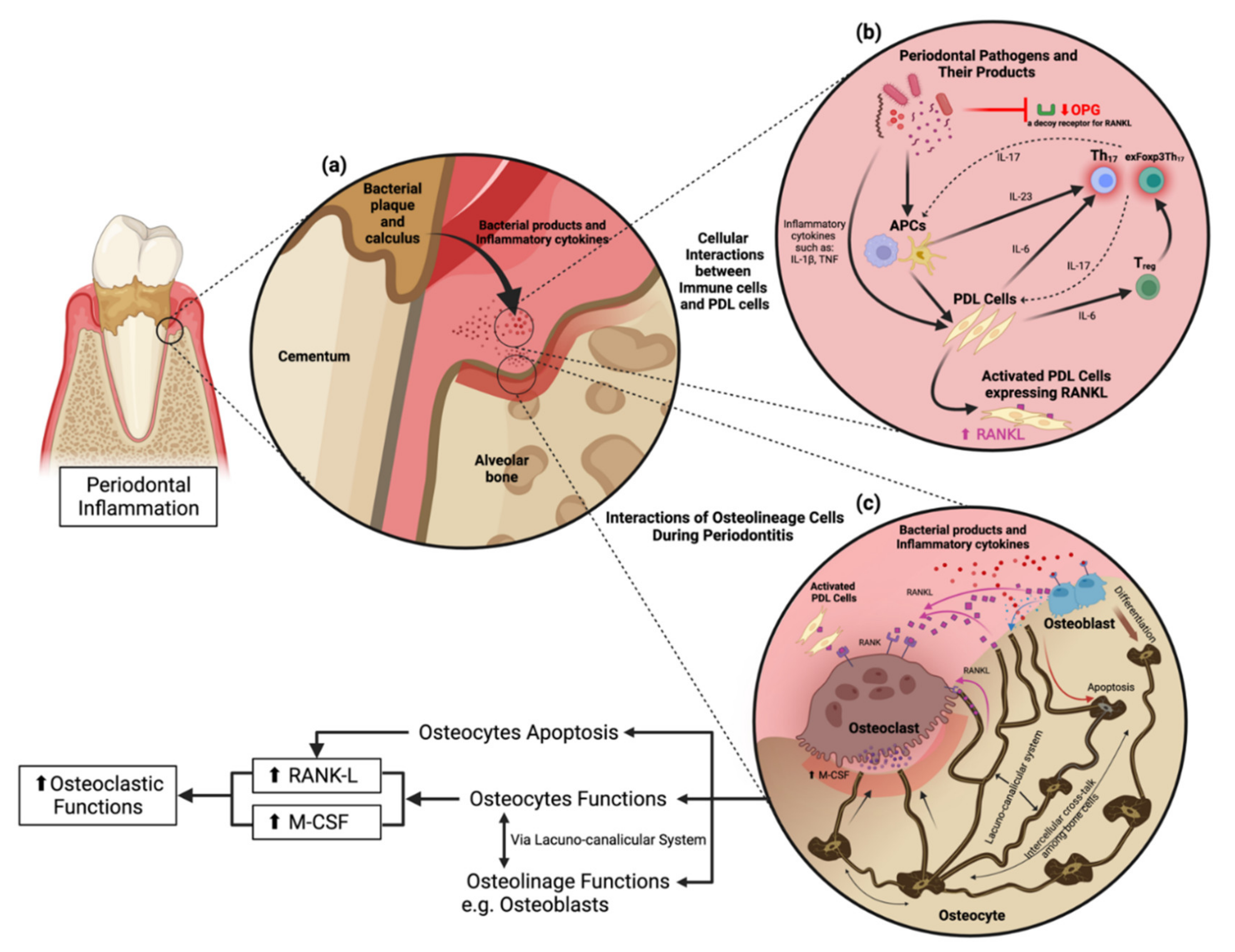
Figure 1. Cellular interactions and osteocyte function in periodontitis. (a) Pathogenic bacteria in the periodontal pocket release their virulence factors to stimulate proinflammatory cytokine production from periodontal stromal cells and immune cells in the periodontium. (b) Cellular interactions in response to periodontitis. Periodontal pathogens and their products activate antigen-presenting cells (APCs) in the periodontium to stimulate periodontal ligament cells (PDL cells) following the activation of Th17 and exFoxp3Th17 cells. (c) Osteocyte functions and their interactions with other cells through the lacunocanalicular system (LCS).
Osteocytes are embedded in the bone matrix, which raises the question of how RANKL produced by osteocytes induces osteoclast differentiation on the bone surface. The precursor of osteoclasts and osteocytes attached within the collagen gel demonstrated that RANKL originates from osteocytes and is delivered to osteoclast precursor mainly via a membrane-bound form employing the dendritic processes of osteocytes in the lacunocanalicular system. In contrast, osteocyte-producing soluble RANKL has a minor role in osteoclastogenesis [51]. Moreover, compressive forces, including orthodontic tooth movement, stimulate osteocyte-mediated osteoclastogenesis through autophagy-mediated RANKL secretion via the transcription factor E3-related signaling [52][53]. Specific removal of osteocyte-derived RANKL inhibits periodontal bone loss in standard and diabetic mice demonstrating that osteocytes play an essential role in the progression of periodontitis [54]. This research has caused a paradigm shift by defining osteocytes as a central RANKL resource.
2.4. Periodontitis Induces Osteocyte Apoptosis Leading to Osteoclastogenesis
Periodontal tissue cells are constantly exposed to microbial pathogens and might undergo cell death [55]. Cell death is the final cellular determination reached after complicated cellular communications and is essential to maintaining homeostasis. The cell death system is generally classified as programmed cell death (PCD) or non-PCD regarding their signal dependency. PCD is divided into two types—apoptotic and non-apoptotic. Apoptosis is cell death characterized by membrane blebbing, cell shrinkage, organelle loss, DNA condensation, and fragmentation [56]. Apoptosis has many purposes, including the elimination of non-functional cells, additional cells, and malignant cells and controlling tissue size, including bone. Osteoblast and osteoclast apoptosis play a role in bone homeostasis [57].
Stimulation of osteocytes with bacteria and inflammatory cytokines induces osteocyte apoptosis in periodontitis. Gingipain, a toxic proteolytic enzyme derived from P. gingivalis, degrades integrin β1 on the cell membrane and suppresses the Rho family of GTPases depolymerizes cytoskeletal protein F-actin leading to reduced cell-cell adhesion to the ECM and osteocyte apoptosis [58]. Moreover, increased proinflammatory cytokines change the osteocyte mechano-sensitivity, induce osteocyte apoptosis, and upregulate the expression of osteocyte-derived inflammatory cytokines and signaling molecules under inflammatory conditions [59].
Osteocyte apoptosis recruits osteoclasts toward apoptosis sites leading to osteoclastogenesis and bone remodeling [60]. The consequence of osteocyte apoptosis contributes to the secretion of several inflammatory cytokines [39][59][61], such as IL-6 [62], resulting in increased RANKL expression in osteocytes [48][49]. Apoptotic osteocytes also release adenosine triphosphate (ATP) via the activated pannexin 1 channel, which acts as the bone lineage cells through ATP receptor gated (P2) channels. This interaction upregulates RANKL expression from nearby surface bone lineage cells to aggregate macrophages to differentiate themselves into osteoclasts [63][64]. Dying osteocytes or apoptotic bodies of osteocytes can directly control osteoclastogenesis and bone remodeling by secreting RANKL [65]. Moreover, apoptotic bodies of osteocytes interact with specific markers on osteoclast precursor cells to promote TNF-α gene expression leading to osteoclastogenesis [66]. The prolonged apoptotic bodies of osteocytes can proceed to secondary apoptosis allowing the cell to secrete various inflammatory cytokines and subsequently activate immune cells to upregulate RANKL expression leading to osteoclastogenesis [67]. The function of the different types of cell death in periodontitis (apoptosis, autophagy, necroptosis, and non-PCD) deserve more exploration. Uncovering the role of apoptosis of osteocytes in the inflammatory condition in periodontitis will lead to a thorough comprehension of periodontitis occurrence and progression, which will support the establishment of future therapeutic prevention, assessment, and management of periodontitis.
2.5. Tissue-Specific Immunity Concept
Recently, the novel concept of “tissue-specific immunity” was introduced to emphasize the roles of local proteins in immunological processes. This theory believes that peripheral tissues and organs are not simply passive immune response targets; instead, they inherently regulate and affect immune function [67][68]. Immune cells require modification to the prerequisites and conditions of the location where these cells are produced or recruited to preserve or rejuvenate their balanced function in tissues. This adaptability regarding the intimate interaction within the tissues is called “functional immune plasticity” [68][69]. Peripheral tissues inherently obtain regulatory activity and produce homeostasis molecules to manipulate the functional plasticity of immune cells to control tissue homeostasis. The discovery of tissue-derived homeostatic molecules as the functional local protein controlling immunological plasticity is critical for comprehension of the internal potential of many organs on immune homeostasis [67]. Studying local regulatory mechanisms and uncovering novel proteins supporting functional immunological plasticity is important to better understand immune-driven inflammatory disorders, such as periodontitis. Consequently, local proteins promoting periodontal regeneration were identified to enable periodontitis treatment without synthetic compounds and antibiotics.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23105540
References
- Kim, H.; Kim, S.; Jung, S. Instruction of microbiome taxonomic profiling based on 16S rRNA sequencing. J. Microbiol. 2020, 58, 193–205.
- Mark Welch, J.L.; Rossetti, B.J.; Rieken, C.W.; Dewhirst, F.E.; Borisy, G.G. Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. USA 2016, 113, E791–E800.
- Nobbs, A.H.; Lamont, R.J.; Jenkinson, H.F. Streptococcus adherence and colonization. Microbiol. Mol. Biol. Rev. 2009, 73, 407–450.
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490.
- Hajishengallis, G.; Lamont, R.J. Beyond the red complex and into more complexity: The polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 2012, 27, 409–419.
- Maekawa, T.; Krauss, J.L.; Abe, T.; Jotwani, R.; Triantafilou, M.; Triantafilou, K.; Hashim, A.; Hoch, S.; Curtis, M.A.; Nussbaum, G.; et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 2014, 15, 768–778.
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759.
- Lamont, R.J.; Hajishengallis, G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol. Med. 2015, 21, 172–183.
- Lenartova, M.; Tesinska, B.; Janatova, T.; Hrebicek, O.; Mysak, J.; Janata, J.; Najmanova, L. The oral microbiome in periodontal health. Front. Cell. Infect. Microbiol. 2021, 11, 119.
- Van Dyke, T.E.; Bartold, P.M.; Reynolds, E.C. The nexus between periodontal inflammation and dysbiosis. Front. Immunol. 2020, 11, 511.
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44.
- Cekici, A.; Kantarci, A.; Hasturk, H.; Van Dyke, T.E. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontology 2000 2014, 64, 57–80.
- Gruber, R. Osteoimmunology: Inflammatory osteolysis and regeneration of the alveolar bone. J. Clin. Periodontol. 2019, 46 (Suppl. S21), 52–69.
- Tsukasaki, M. RANKL and osteoimmunology in periodontitis. J. Bone Miner. Metab. 2021, 39, 82–90.
- Tsukasaki, M.; Komatsu, N.; Nagashima, K.; Nitta, T.; Pluemsakunthai, W.; Shukunami, C.; Iwakura, Y.; Nakashima, T.; Okamoto, K.; Takayanagi, H. Host Defense against oral microbiota by bone-damaging T Cells. Nat. Commun. 2018, 9, 701.
- Yoshida, H.; Hayashi, S.; Kunisada, T.; Ogawa, M.; Nishikawa, S.; Okamura, H.; Sudo, T.; Shultz, L.D.; Nishikawa, S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990, 345, 442–444.
- Takahashi, K.; Azuma, T.; Motohira, H.; Kinane, D.F.; Kitetsu, S. The potential role of interleukin-17 in the immunopathology of periodontal disease. J. Clin. Periodontol. 2005, 32, 369–374.
- Maekawa, T.; Briones, R.A.; Resuello, R.R.; Tuplano, J.V.; Hajishengallis, E.; Kajikawa, T.; Koutsogiannaki, S.; Garcia, C.A.; Ricklin, D.; Lambris, J.D.; et al. Inhibition of pre-existing natural periodontitis in non-human primates by a locally administered peptide inhibitor of complement C3. J. Clin. Periodontol. 2016, 43, 238–249.
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179.
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Lüthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319.
- López Roldán, A.; García Giménez, J.L.; Alpiste Illueca, F. Impact of periodontal treatment on the RANKL/OPG ratio in crevicular fluid. PLoS ONE 2020, 15, e0227757.
- Maekawa, T.; Abe, T.; Hajishengallis, E.; Hosur, K.B.; DeAngelis, R.A.; Ricklin, D.; Lambris, J.D.; Hajishengallis, G. Genetic and intervention studies implicating complement C3 as a major target for the treatment of periodontitis. J. Immunol. 2014, 192, 6020–6027.
- Xiong, J.; Cawley, K.; Piemontese, M.; Fujiwara, Y.; Zhao, H.; Goellner, J.J.; O’Brien, C.A. Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat. Commun. 2018, 9, 2909.
- Asano, T.; Okamoto, K.; Nakai, Y.; Tsutsumi, M.; Muro, R.; Suematsu, A.; Hashimoto, K.; Okamura, T.; Ehata, S.; Nitta, T.; et al. Soluble RANKL is physiologically dispensable but accelerates tumour metastasis to bone. Nat. Metab. 2019, 1, 868–875.
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL Biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 2.
- Crotti, T.; Smith, M.D.; Hirsch, R.; Soukoulis, S.; Weedon, H.; Capone, M.; Ahern, M.J.; Haynes, D. Receptor activator NF kappaB ligand (RANKL) and osteoprotegerin (OPG) protein expression in periodontitis. J. Periodontal Res. 2003, 38, 380–387.
- Ochiai, N.; Nakachi, Y.; Yokoo, T.; Ichihara, T.; Eriksson, T.; Yonemoto, Y.; Kato, T.; Ogata, H.; Fujimoto, N.; Kobayashi, Y.; et al. Murine osteoclasts secrete serine protease HtrA1 capable of degrading osteoprotegerin in the bone microenvironment. Commun. Biol. 2019, 2, 86.
- Akiyama, T.; Miyamoto, Y.; Yoshimura, K.; Yamada, A.; Takami, M.; Suzawa, T.; Hoshino, M.; Imamura, T.; Akiyama, C.; Yasuhara, R.; et al. Porphyromonas gingivalis-derived lysine gingipain enhances osteoclast differentiation induced by tumor necrosis factor-alpha and interleukin-1 beta but suppresses that by interleukin-17A: Importance of proteolytic degradation of osteoprotegerin by lysine gingipain. J. Biol. Chem. 2014, 289, 15621–15630.
- Huang, X.; Xie, M.; Xie, Y.; Mei, F.; Lu, X.; Li, X.; Chen, L. The roles of osteocytes in alveolar bone destruction in periodontitis. J. Transl. Med. 2020, 18, 479.
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238.
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 2006, 235, 176–190.
- Bonewald, L.F. Osteocytes as dynamic multifunctional cells. Ann. N. Y. Acad. Sci. 2007, 1116, 281–290.
- Bellido, T. Osteocyte-driven bone remodeling. Calcif. Tissue Int. 2014, 94, 25–34.
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J. Bone Miner. Metab. 2021, 39, 19–26.
- Koide, M.; Kobayashi, Y. Regulatory mechanisms of sclerostin expression during bone remodeling. J. Bone Miner. Metab. 2019, 37, 9–17.
- Hienz, S.A.; Paliwal, S.; Ivanovski, S. Mechanisms of bone resorption in periodontitis. J. Immunol. Res. 2015, 2015, 615486.
- Cassuto, J.; Folestad, A.; Göthlin, J.; Malchau, H.; Kärrholm, J. The key role of proinflammatory cytokines, matrix proteins, RANKL/OPG and Wnt/beta-catenin in bone healing of hip arthroplasty patients. Bone 2018, 107, 66–77.
- Harris, S.E.; MacDougall, M.; Horn, D.; Woodruff, K.; Zimmer, S.N.; Rebel, V.I.; Fajardo, R.; Feng, J.Q.; Gluhak-Heinrich, J.; Harris, M.A.; et al. Meox2Cre-mediated disruption of CSF-1 leads to osteopetrosis and osteocyte defects. Bone 2012, 50, 42–53.
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-related cytokines regulate osteoclast formation and bone resorption. Int. J. Mol. Sci. 2020, 21, 5169.
- Werner, S.L.; Sharma, R.; Woodruff, K.; Horn, D.; Harris, S.E.; Gorin, Y.; Lee, D.Y.; Hua, R.; Gu, S.; Fajardo, R.J.; et al. CSF-1 in osteocytes inhibits Nox4-mediated oxidative stress and promotes normal bone homeostasis. JBMR Plus 2020, 4, e10080.
- Liu, D.; Xu, J.K.; Figliomeni, L.; Huang, L.; Pavlos, N.J.; Rogers, M.; Tan, A.; Price, P.; Zheng, M.H. Expression of RANKL and OPG mRNA in periodontal disease: Possible involvement in bone destruction. Int. J. Mol. Med. 2003, 11, 17–21.
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602.
- Theoleyre, S.; Wittrant, Y.; Tat, S.K.; Fortun, Y.; Redini, F.; Heymann, D. The molecular triad OPG/RANK/RANKL: Involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004, 15, 457–475.
- Martin, T.J.; Sims, N.A. RANKL/OPG; Critical role in bone physiology. Rev. Endocr. Metab. Disord. 2015, 16, 131–139.
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234.
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241.
- Sakamoto, E.; Kido, J.I.; Takagi, R.; Inagaki, Y.; Naruishi, K.; Nagata, T.; Yumoto, H. Advanced glycation end-product 2 and Porphyromonas gingivalis lipopolysaccharide increase sclerostin expression in mouse osteocyte-like cells. Bone 2019, 122, 22–30.
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 Receptor system and its role in physiological and pathological conditions. Clin. Sci. 2012, 122, 143–159.
- Yu, K.; Ma, Y.; Li, X.; Wu, X.; Liu, W.; Li, X.; Shen, J.; Wang, H. Lipopolysaccharide increases IL-6 secretion via activation of the ERK1/2 signaling pathway to upregulate RANKL gene expression in MLO-Y4 cells. Cell Biol. Int. 2017, 41, 84–92.
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-alpha directly enhances osteocyte RANKL expression and promotes osteoclast formation. Front. Immunol. 2019, 10, 2925.
- Honma, M.; Ikebuchi, Y.; Kariya, Y.; Hayashi, M.; Hayashi, N.; Aoki, S.; Suzuki, H. RANKL subcellular trafficking and regulatory mechanisms in osteocytes. J. Bone Miner. Res. 2013, 28, 1936–1949.
- Shoji-Matsunaga, A.; Ono, T.; Hayashi, M.; Takayanagi, H.; Moriyama, K.; Nakashima, T. Osteocyte regulation of orthodontic force-mediated tooth movement via RANKL expression. Sci. Rep. 2017, 7, 8753.
- Li, W.; Zhao, J.; Sun, W.; Wang, H.; Pan, Y.; Wang, L.; Zhang, W.B. Osteocytes promote osteoclastogenesis via autophagy-mediated RANKL secretion under mechanical compressive force. Arch. Biochem. Biophys. 2020, 694, 108594.
- Graves, D.T.; Alshabab, A.; Albiero, M.L.; Mattos, M.; Corrêa, J.D.; Chen, S.; Yang, Y. Osteocytes play an important role in experimental periodontitis in healthy and diabetic mice through expression of RANKL. J. Clin. Periodontol. 2018, 45, 285–292.
- Jun, H.K.; Jung, Y.J.; Choi, B.K. Treponema denticola, Porphyromonas gingivalis, and Tannerella forsythia induce cell death and release of endogenous danger signals. Arch. Oral Biol. 2017, 73, 72–78.
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features . World Acad. Sci. J. 2020, 2, 39–48.
- Jilka, R.L.; Noble, B.; Weinstein, R.S. Osteocyte apoptosis. Bone 2013, 54, 264–271.
- Qiu, Q.; Zhang, F.; Wu, J.; Xu, N.; Liang, M. Gingipains disrupt F-actin and cause osteoblast apoptosis via integrin beta1. J. Periodontal Res. 2018, 53, 762–776.
- Zhou, M.; Li, S.; Pathak, J.L. Pro-inflammatory cytokines and osteocytes. Curr. Osteoporos. Rep. 2019, 17, 97–104.
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The osteocyte: An endocrine cell … and more. Endocr. Rev. 2013, 34, 658–690.
- Rose-John, S. The soluble interleukin 6 receptor: Advanced therapeutic options in inflammation. Clin. Pharmacol. Ther. 2017, 102, 591–598.
- Cheung, W.Y.; Simmons, C.A.; You, L. Osteocyte apoptosis regulates osteoclast precursor adhesion via osteocytic IL-6 secretion and endothelial ICAM-1 expression. Bone 2012, 50, 104–110.
- McCutcheon, S.; Majeska, R.J.; Spray, D.C.; Schaffler, M.B.; Vazquez, M. Apoptotic osteocytes induce RANKL production in bystanders via purinergic signaling and activation of pannexin channels. J. Bone Miner. Res. 2020, 35, 966–977.
- Komori, T. Cell death in chondrocytes, osteoblasts, and osteocytes. Int. J. Mol. Sci. 2016, 17, 2045.
- Al-Dujaili, S.A.; Lau, E.; Al-Dujaili, H.; Tsang, K.; Guenther, A.; You, L. Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J. Cell. Biochem. 2011, 112, 2412–2423.
- Algate, K.; Haynes, D.R.; Bartold, P.M.; Crotti, T.N.; Cantley, M.D. The effects of tumour necrosis factor-alpha on bone cells involved in periodontal alveolar bone loss; Osteoclasts, osteoblasts and osteocytes. J. Periodontal Res. 2016, 51, 549–566.
- Hajishengallis, G.; Chavakis, T. DEL-1-regulated immune plasticity and inflammatory disorders. Trends Mol. Med. 2019, 25, 444–459.
- Galli, S.J.; Borregaard, N.; Wynn, T.A. Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 2011, 12, 1035–1044.
- Oyler-Yaniv, A.; Oyler-Yaniv, J.; Whitlock, B.M.; Liu, Z.; Germain, R.N.; Huse, M.; Altan-Bonnet, G.; Krichevsky, O. A tunable diffusion-consumption mechanism of cytokine propagation enables plasticity in cell-to-cell communication in the immune system. Immunity 2017, 46, 609–620.
This entry is offline, you can click here to edit this entry!