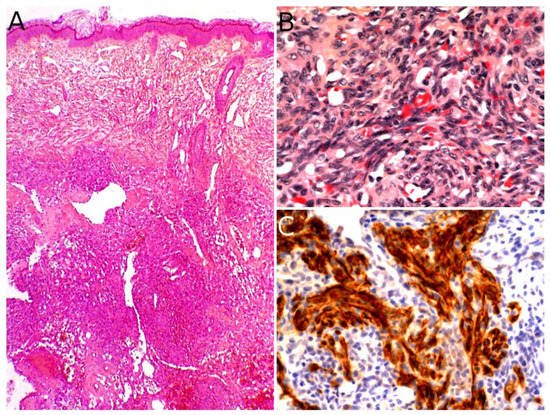
The diagnosis rests on the pathological analysis of a biopsy (including fine-needle biopsy), which should be as deep as possible. Indeed, a superficial sampling of the tumor may show misleading aspects, such as lipofibromatosis-like [
11], hemangioma-like features [
10,
12] or fat necrosis. This can be avoided by favoring a deep surgical biopsy or fine needle biopsy (
Figure 3B,C). More often than not, infantile fibrosarcoma demonstrates a cellular, monomorphic neoplasm made up of ovoid to spindle cells arranged in compact sheets or long fascicles, sometimes assuming the typical herringbone pattern (
Figure 3D). Mitoses can be sparse or numerous from one tumor to another and in the same tumor, with no prognostic impact. Necrosis is possible and has no prognostic implication either. The tumor vessels may be numerous. In such cases, the tumor might even be misdiagnosed as a vascular anomaly. The immunophenotype is not specific. The tumor may show variable and focal expression of SMA, S100, CD34, and rarely desmin (or none) [
13]. Expression of pan-TRK (neurotrophic tyrosine receptor kinase) is helpful for screening tumors with a
NTRK gene rearrangement [
14]. Indeed, most infantile fibrosarcomas exhibit an
ETV6-NTRK3 gene fusion resulting from the t(12;15)(p13;q25) chromosomal translocation. Other
NTRK3 fusion partners include
EML4 [
15],
RBPMS [
16], or
SPECC1L [
17]. Other tyrosine kinase genes are involved in fusions, including
NTRK1,
NTRK2,
RET,
MET, and
RAF1, with a variety of partners [
16,
18,
19,
20].
BRAF fusions, complex deletions, and point mutations have also been evidenced, inducing constitutive activation of the RAF-MEK-ERK pathway [
18,
19]. Interestingly, other spindle cell tumors harboring
RAF1,
BRAF, and
NTRK1/2 fusions and further characterized by S100 and CD34 co-expression (discussed hereafter, see
Section 2.3), demonstrate a close proximity to some infantile fibrosarcomas by RNA unsupervised hierarchical clustering analysis [
21]. This suggests a common pathogenesis between these tumors, or even a common spectrum of low-grade spindle cell tumors with activation of the RAF-MEK-ERK pathway.
Indeed,
NTRK-rearranged spindle cell neoplasms include a wide variety of morphologies with tumors described as infantile fibrosarcoma-like, inflammatory myofibroblastic tumor-like (IMT-like), lipofibromatosis-like neural tumor (LPF-NT), and MPNST-like [
36]. Microscopically, the most consistent features are an infiltrative growth pattern within subcutaneous fat, dense spindle cells haphazardly arranged or in fascicles, elongated nuclei with mild atypia and hyperchromasia, inconspicuous nucleoli, and low mitotic activity (
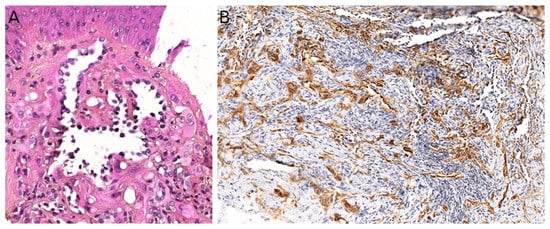
Figure 4A,B) [
16,
21,
33,
34,
35,
36]. A prominent inflammatory infiltrate has been described in IMT-like cases [
16,
21,
34,
37]. The clue to the diagnosis is the co-expression of CD34 and S100 by immunohistochemistry. The expression may be focal and vary among cases [
34,
35]. The retained expression of H3K27me3 can help in the differential diagnosis with a MPNST, a differential that may be considered only in adolescents. Screening with an anti-NTRK1 or panTRK antibody is also helpful (
Figure 4C). The definitive diagnosis is made by molecular testing for
NTRK1 gene fusions (in most cases,
Figure 4D). The fusion partners include
LMNA,
TPM3,
TPR, and
SQSTM1 [
16,
34]. Other gene rearrangements have been identified in children, involving
NTRK3 with
EML4 or
KHDRBS1 as partners [
21,
38,
39]. We have already discussed the possible relation between
NTRK-rearranged spindle cell neoplasms and infantile fibrosarcoma (see
Section 2.2).
3. Vascular Tumors
3.1. Kaposiform Hemangioendothelioma
The incidence of kaposiform hemangioendothelioma (KHE) is probably under evaluated. It has been suggested that small asymptomatic or atypical KHE may be misdiagnosed as variants of other vascular tumors [
76]. Although the exact incidence and prevalence of this vascular tumor are not known, it is clear that there is a peak within the first year of life. Around 50% of superficial cases are congenital [
76]. It is admitted now that KHE and tufted angioma belong to the same spectrum and that they share the same biology and probably the same pathogenesis [
77,
78]. Actually, some specialists argue that KHE might only be a florid presentation of tufted angioma, and that the adverse clinical course of this tumor is purely dependent on the occurrence of Kasabach–Merritt phenomenon (KMP) or vital organ compression. For this reason, KHE may well be benign, the morbidity and mortality being linked to secondary complications. We decided to include KHE in this review anyway, but this decision is debatable and it will not be discussed extensively.
KHE usually involves the deep dermis and subcutis, but deeper lesions are possible, with no cutaneous signs in about 12% of cases [
76,
78]. There is a slightly higher prevalence in the limbs, but all sites are possible. Most of the time, the tumor is unique, and can adopt many clinical aspects: erythematous sometimes purple; papules, nodules or plaques or an indurated mass; with varying degrees of infiltration [
78]. It is a big, rapidly growing tumor, 3 to 27 cm in greater dimension [
79]. One of the most severe complications of KHE, also seen in tufted angioma, is the KMP, a life-threatening consumptive coagulopathy with severe thrombocytopenia. It occurs in 42 to 71% of cases [
80,
81,
82]. There is a higher frequency of KMP in congenital, large KHE, especially above 8 cm [
83]. In cases of KMP, the KHE lesions appear purpuric, congestive, and painful [
84]. Spontaneous hemorrhage is rare but any invasive procedure (biopsy, excision), trauma, or ulceration may lead to significant bleeding. Other complications of KHE include musculoskeletal disorders due to tumor infiltration, lymphedema, and compression of vital structures (e.g., airway obstruction in a KHE of the neck) [
78].
US reveals an ill-defined, heterogeneous lesion, most often hyperechoic [
79]. On color Doppler US, the tumor is hypervascular. MRI reveals a heterogeneous, hyperintense appearance on T2-weighted images, with heterogeneous, generally intense enhancement [
79]. Overall, the imaging findings lack specificity and are characterized by a wide range of aspects regarding the degree of infiltration, signal intensities, and enhancement patterns [
79].
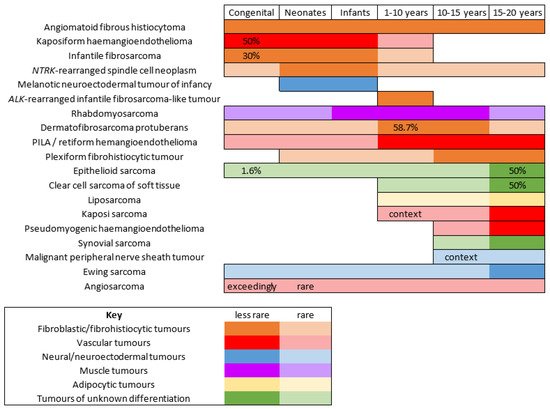
The gold standard for diagnosis is the biopsy. KHE is a deep dermal and subcutaneous, infiltrating proliferation of spindle endothelial cells arranged in nodules (
Figure 7A). The tumor can adopt a “cannonball” pattern, especially in the dermis, similar to a tufted angioma, but the nodules are larger, less defined, and coalescent. The tumor is also more infiltrative. Mitoses are rare and not abnormal (
Figure 7B) [
78,
80,
85]. Most of the spindle cells are positive for lymphatic markers podoplanin, LYVE1, and PROX1 (
Figure 7C) [
80,
86].
Figure 7. Kaposiform hemangioendothelioma: (A) Deep dermal and subcutaneous, infiltrating proliferation of spindle cells arranged in nodules (×25); (B) spindle endothelial cells with no mitoses (×200); (C) positivity for the lymphatic marker podoplanin in the spindle cells (×200).
Similar to other vascular anomalies, numerous mutations involving G proteins have been found in KHE and tufted angioma, especially in the
GNA14 gene, a paralogue of
GNAQ [
87]. These genes encode the Gα subunit that, along with Gβ and Gγ in a heterotrimer, binds G protein-coupled receptors.
The prognosis depends on the size and location of the tumor. Large, deep KHE tumors are associated with higher morbidity and mortality due to tumor infiltration, compression of vital organs, and KMP [
78]. Treatment with corticosteroids and/or vincristine has been recommended but is based on expert opinion and lack a well-designed clinical study [
84]. Wide surgical excision can be curative but is rarely feasible. For a decade now, sirolimus has been emerging as a treatment option. The first report of a successful treatment in a child was published in 2010 [
88]. Some of the concerns with sirolimus have been the occurrence of adverse events and the issue of the right dose in the pediatric population [
89]. Recently, recommendations for the initial dose in children were proposed. Due to sirolimus being metabolized by cytochrome P450 3A (CYP3A), the initial dose depends both on the weight and the
CYP3A5 genotype [
90]. A recent prospective study of sirolimus for complicated vascular anomalies underlined the importance of closely monitoring possible adverse events, while confirming the effectiveness of the treatment [
91]. In this study, the most frequent adverse events (>20% of cases) were mucositis, upper respiratory infections, and nausea/vomiting.
3.2. Papillary Intralymphatic Angioendothelioma (PILA)/Retiform Hemangioendothelioma
Papillary intralymphatic angioendothelioma (PILA, also known as Dabska tumor) and retiform hemangioendothelioma belong to the same spectrum. These two rare tumors are lymphatic in origin. Cases with overlapping features of PILA and retiform hemangioendothelioma have been described, which is the reason why some authors have suggested to include both tumors under the name
hobnail hemangioendothelioma [
92]. However, pure forms of retiform hemangioendothelioma are very rare in children; PILA or mixed tumors are much more frequent. PILA usually involves the dermis and subcutaneous tissues of the head and neck in children and adolescents, whereas retiform hemangioendothelioma more often involves the upper and lower limbs of young adults and adolescents, with some reported cases in the head or trunk [
93]. Similar to Masson’s tumor, PILA has been reported to arise in a pre-existing vascular malformation, but also on chronic lymphedema and in intramuscular hemangiomas. There are a few reports of a retiform hemangioendothelioma arising on a pre-existing lymphatic malformation [
94].
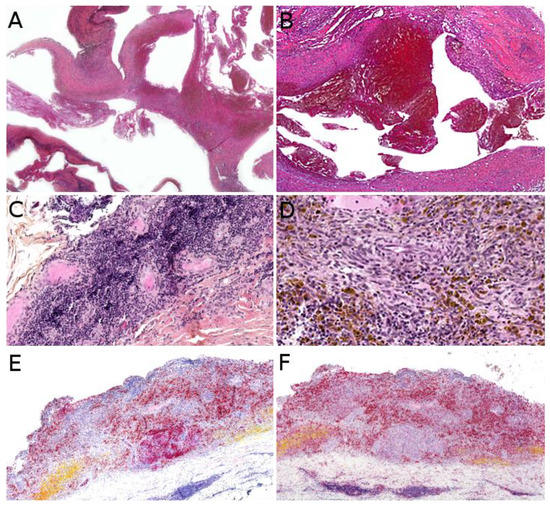
PILA and retiform hemangioendothelioma both present as a slowly growing, firm, solitary nodule or mass, more rarely plaque; all nonspecific findings, which explains the diagnosis not being raised by dermatologists or pediatricians. To the best of our knowledge, there has been no study of the imaging characteristics of these tumors in the English literature.
Microscopically, PILA consists of vessels of various sizes, more or less dilated, containing papillary tufts protruding from the vessel wall and sometimes occluding the lumen. The papillae are centered by a hyalinized core and lined by hobnail endothelial cells with large hyperchromatic nuclei located towards the luminal pole of the cell (
Figure 8A). Mitoses are sparse. Retiform hemangioendothelioma involves the whole dermis and often infiltrates the subcutaneous tissue. It is made of elongated, anastomosing vessels reminiscent of the architecture of the rete testis. In both PILA and retiform hemangioendothelioma, there is often a prominent lymphocytic infiltrate. The immunophenotype is similar in both tumors, with positivity for CD31 and CD34 in the endothelial cells. Prox1 and podoplanin (D2-40) are positive in most cases of PILA (
Figure 8B) [
95,
96,
97,
98]. Of note, rare cases of composite hemangioendothelioma have been described in adolescents, consisting of areas of retiform hemangioendothelioma and solid areas with a vaguely neuroendocrine morphology. These cases showed no recurrence nor metastasis in children, however, both of these have been reported in adults, especially bone and lung metastases [
96]. PILA has intermediate malignant potential with possible local recurrences and exceptional lymph node metastases. Lung metastases have been described in at least one case [
99]. Retiform hemangioendothelioma shares a similar clinical course.
Figure 8. Papillary intralymphatic angioendothelioma: (A) Vascular proliferation with papillary tufts protruding from the vessel wall, lined by hobnail endothelial cells with large hyperchromatic nuclei (×200); (B) partial positivity for the lymphatic marker podoplanin (×100).
The treatment of choice of PILA and retiform hemangioendothelioma is complete surgical resection, in order to avoid recurrences. However, clear surgical margins may be hard to achieve in some cases of retiform hemangioendothelioma, due to its infiltrative growth pattern. In such cases, MMS may be useful, as reported in a 11-year-old girl with a tumor of the finger [
100]. In unresectable cases, association of radiation therapy and chemotherapy with cisplatin might be an option, as reported in an adult [
101].
3.3. Pseudomyogenic Haemangioendothelioma
Pseudomyogenic hemangioendothelioma (PMHE) is a rare vascular tumor of intermediate malignant potential. It affects adolescents and young adults, with a mean age at diagnosis close to 30 years and a strong male predominance (sex ratio about 4:1) [
102,
103]. The most frequently involved sites are the extremities, especially the lower limbs, followed by the trunk and upper limbs, but the tumor has been described in other locations. PMHE arises mainly in the soft tissues but may occur in the dermis, muscle, or bone [
102]. A characteristic feature of this tumor is the frequent involvement of different tissue planes, resulting in a mass or deep-seated nodule, with pain in about half of cases. Multicentric presentation is also common [
102,
104,
105]. On MRI, PMHE is hypointense on T1-weighted images and hyperintense on T2-weighted and stir-weighted images. It has been suggested that positron emission tomography scan (PET-scan) might be helpful to reveal possible occult deep lesions [
8].
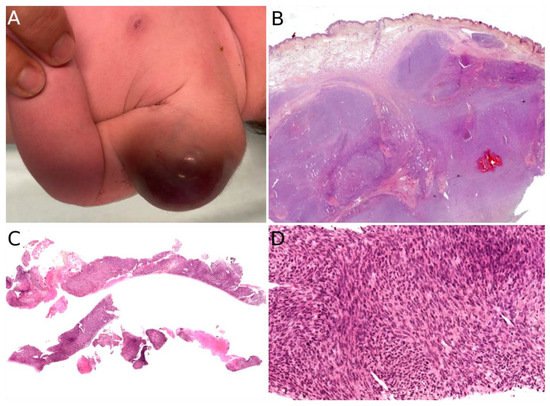
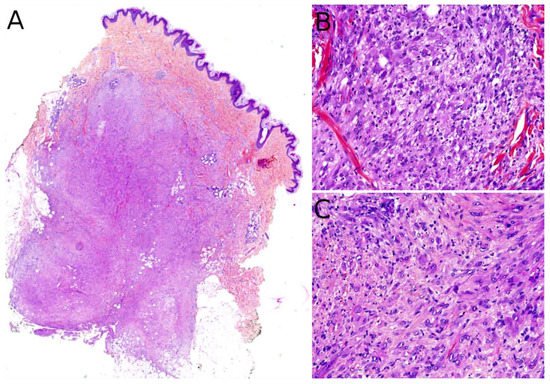
Grossly, PMHE is usually made of multifocal, ill-defined, 1 to 3 cm large, white- or brown-colored nodules [
105]. Microscopically, PMHE has an infiltrative pattern of growth (
Figure 9A). It is made of sheets and fascicles of epithelioid cells or plump spindle cells with a deeply eosinophilic cytoplasm which can be reminiscent of rhabdomyoblasts (
Figure 9B). The stroma is loose, rarely myxoid, and contains numerous neutrophils in about half of cases (
Figure 9C) [
105]. Similar to dermatofibroma, epidermal hyperplasia may be present above the tumor [
106]. Intravascular invasion has been described but does not seem to have any prognostic impact [
107]. PMHE co-expresses cytokeratins and endothelial markers. It is diffusely positive for cytokeratin AE1/AE3 and ERG. CD31 is positive in about half of cases. SMA may be expressed focally in some cases. Interestingly, there is diffuse nuclear positivity for FOSB in 96% of cases, a very helpful finding since FOSB is expressed in only a couple of other vascular tumors: epithelioid hemangioma (54% of cases), which is not a histological differential diagnosis of PMHE, and some rare cases of epithelioid angiosarcoma (5%), spindle-cell angiosarcoma (10%), or epithelioid hemangioendothelioma (5%) [
104]. One of the main differential diagnoses of PMHE is epithelioid sarcoma. However, INI1 is normally expressed in PMHE.
Figure 9. Pseudomyogenic hemangioendothelioma: (A) Dense cellular proliferation with an infiltrating pattern of growth (×25); (B) sheets of epithelioid cells or plump spindle cells with a deeply eosinophilic cytoplasm (×100); (C) rhabdomyoblast-like cells with moderate nuclear atypia and some neutrophils in the stroma (×200).
FOSB positivity is related to the presence of the t(7;19)(q22;q13) translocation responsible for a
SERPINE1-FOSB gene fusion in the majority of PMHE [
102,
108]. Recently, other partners for
FOSB have been found in some cases of PMHE: the beta-actin gene, responsible for the
ACTB-FOSB gene fusion [
109,
110]; the WW domain-containing transcription regulator 1 gene, responsible for the
WWTR1-FOSB gene fusion in an isolated case [
111]; and more recently, the clathrin heavy chain gene, responsible for the
CLTC-FOSB gene fusion [
112]; and the epidermal growth factor-like 7 gene, responsible for the
EGFL7-FOSB gene fusion [
113]. It remains to be determined if these various gene fusions are associated with different clinicopathological features. To date, there is no targeted therapy for
FOSB gene fusions.
The clinical course is characterized by local recurrences in about 60% of cases, often multiple and with possible additional nodules. Regional lymph node metastasis has been described in only one patient, and distant metastases in three patients; only two patients died of disease [
107,
114,
115]. The treatment of choice is complete surgical excision. Some reports of clinical improvement following treatment with sirolimus or everolimus (anti-mTOR) are hopeful for non resectable cases [
116,
117].
3.4. Angiosarcoma
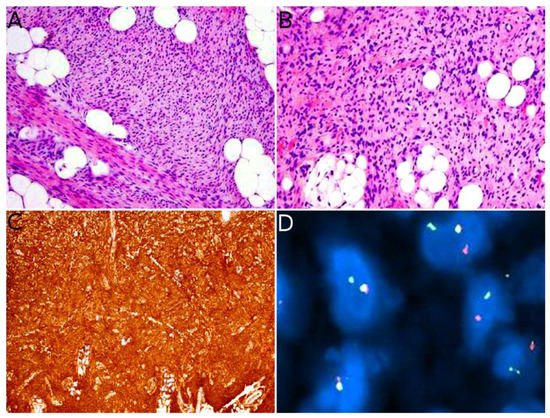
Angiosarcoma is extremely rare in children, therefore, it is impossible to draw reliable epidemiological or clinical data on angiosarcoma in children and in the skin. In children, it is mostly seen in association with genetic conditions such as xeroderma pigmentosum, or Aicardi syndrome, or after radiation therapy [
85]. Therefore, angiosarcomas are almost always seen in older children and adolescents. Congenital or infantile cases are exceedingly rare. Angiosarcomas in children seem to predominate in girls and in the lower extremities, but these observations should be tempered since they are based on a very small series of 10 cases [
118]. The tumor is often a rapidly enlarging nodule or mass, sometimes ulcerated (
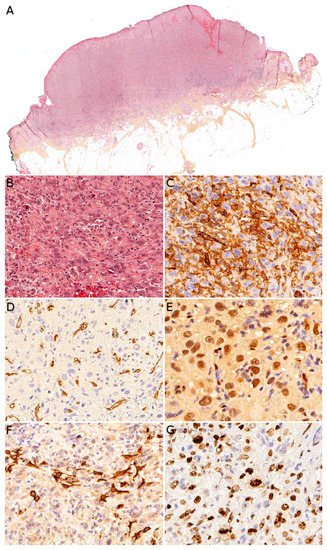
Figure 10A). It may be centered in the dermis, in the subcutaneous tissue, or in both. The tumor is made of a branching network of vessels admixed with more solid areas made of epithelioid cells (
Figure 10B). Indeed, the epithelioid variant is particularly frequent in children (90%). Nuclear atypia is often prominent, at least in some areas. Mitotic activity is high in most cases (
Figure 10G), but may be low. Necrosis is possible. Immunohistochemistry reveals a positivity for at least one endothelial marker, with a better sensitivity of CD31 and ERG as compared with CD34 (
Figure 10C–E) [
118]. Podoplanin is commonly positive. HHV8 is negative. Aberrant cytokeratin positivity is possible (
Figure 10F). Wide surgical excision is the treatment of choice. Adjuvant chemotherapy may be useful but reported cases are too few to draw any recommendation and each case should be discussed collegially.
Figure 10. Angiosarcoma: (A) Ulcerated nodule (×25); (B) atypical epithelioid cells with numerous mitoses and a tripolar mitosis (center of the panel) (×200); (C) strong CD31 positivity (×200); (D) partial CD34 positivity on small dystrophic vascular structures (×200); (E) strong nuclear ERG positivity (×200); (F) partial positivity for the cytokeratin marker AE1/AE3 (×200); (G) high proliferation index and multiple mitoses (Ki67 ×200).
4. Muscular Tumors: Rhabdomyosarcomas
Primary cutaneous rhabdomyosarcoma (RMS) is a rare tumor, with about 55 cases reported in the literature [
125,
126]. It occurs mainly in the head and neck, and there seem to be no sex predominance [
125]. The age range is wide with rare congenital/neonatal cases, but also cases in older children and adolescents. Importantly, the two main histological subtypes of RMS, embryonal RMS (ERMS) and alveolar RMS (ARMS), have distinct clinical and histological characteristics. Therefore, they are discussed separately hereafter. Contrary to non-cutaneous RMS, where ERMS account for nearly 75% of all RMS, primarily cutaneous RMS are more often of the alveolar subtype [
125], even in congenital/neonatal cases. Indeed, in the skin and superficial soft tissues, after a short review of the literature, we found that congenital/neonatal ARMS were twice as often represented as congenital/neonatal ERMS (20 ARMS, 9 ERMS) [
127,
128,
129,
130,
131,
132]. In children, tumors with extensive pleomorphism should be considered anaplastic ERMS and not pleomorphic RMS [
133]. To the best of our knowledge, no pediatric primary cutaneous spindle-cell/sclerosing RMS has been reported in the literature. Still, it seems important to keep in mind this differential diagnosis when confronted with a spindle-cell tumor, since this RMS subtype is associated with a better prognosis and indolent clinical course.
NCOA2 and
VGLL2 rearrangements are the most common in spindle-cell/sclerosing RMS in children and should be sought for establishing this diagnosis [
134].
4.1. Alveolar Rhabdomyosarcoma (ARMS)
Congenital/neonatal ARMS has a slight female predominance [
130,
135]. Although very rare in this age group, congenital/neonatal ARMS has other particularities which are worth mentioning: the tumor occurs mainly in the limbs, followed by the head and neck, and the trunk [
130]; according to a recent review, cutaneous and subcutaneous locations are most often metastatic sites [
130,
136] however, at least four reported cases of congenital ARMS were primarily cutaneous [
128,
131,
135,
137]. All ages considered, ARMS is usually an erythematous mass or nodule, initially misdiagnosed as a hemangioma, but often rapidly growing, ranging from 4 to 12 cm in greater diameter [
128,
131,
137]. When congenital/neonatal, it may be multiple, even in primarily cutaneous cases. In older children, the lesion is unique (
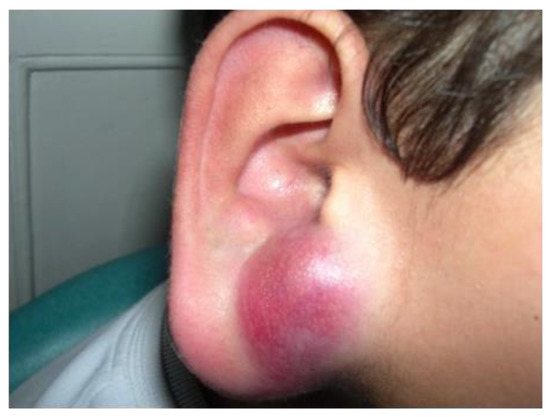
Figure 12).
Figure 12. Rhabdomyosarcoma of the ear in a young boy: solitary erythematous nodule.
Microscopically, primarily cutaneous ARMS is similar to ARMS in other locations; it is composed of nests of undifferentiated small round cells with a high N/C ratio and a variable proportion of tumor cells with a moderate eosinophilic cytoplasm. True differentiated rhabdomyoblasts are rare. In the center of the nests, the cells show discohesion with a tendency to disaggregate, leading to the alveolar pattern. Multinucleated tumor cells are a rather common finding. Mitoses can be sparse or more prominent and necrosis is possible [
127,
128,
132]. Tumor cells express skeletal muscle markers: desmin, myogenin (Myf4), and/or MyoD1.
The overall prognosis of congenital ARMS is poor, with about 75% of deaths in the review by Russo et al. However, about 75% of cases in this review also had metastases, with 41% of central nervous system metastases [
130]. It is possible that primary cutaneous cases of congenital ARMS have a better prognosis, as reported [
128,
131,
135,
137]. Importantly, the prognosis of RMS, whatever the age of the child, has been linked to its genetic anomalies; patients with
FOXO1 fusion positive RMS having a worse outcome than patients with
FOXO1 fusion negative RMS, and the
PAX7-FOXO1 fusion has a better prognosis than the
PAX3-FOXO1 fusion [
138]. Since 70% of ARMS show either one of the
FOXO1 fusions, it makes sense that ARMS should have a poorer prognosis than ERMS. However, in congenital/neonatal ARMS cases, only three
FOXO1 fusions have been reported, to our knowledge, whereas the majority of cases seem to be negative for
FOXO1 fusions [
127,
129,
130,
135]. This suggests a possible different pathogenesis for congenital/neonatal ARMS. For instance, some cases have been linked to Beckwith–Wiedemann syndrome and the tumors seem to arise from silencing of the
CDKN1C gene through epigenetic mechanisms in this syndrome [
129,
135].
4.2. Embryonal Rhabdomyosarcoma (ERMS)
Congenital/neonatal ERMS mainly occurs in the head and neck, followed by the trunk. The male/female ratio is about two [
130]. Only nine cases of congenital/neonatal ERMS of the skin or superficial soft tissues have been reported, to the best of our knowledge, with fragmentary data, which makes it difficult to draw meaningful conclusions [
127,
139,
140,
141]. The locations include the cheek, scalp, neck, hand, foot, and lower back. All ages considered, the tumor is firm, ranges from 3 to 15 cm in greater dimension, may be ulcerated if large, with possible associated lymphadenopathy [
127,
139,
141].
Microscopically, ERMS shows alternating loose and dense cellularity in a variably myxoid stroma. The tumor cells range from stellate to small round undifferentiated to spindle. Unlike ARMS, myogenin expression is more variable.
The prognosis is deemed to be better than that of ARMS, however, in congenital/neonatal cases at least three out of nine ERMS cases were fatal after only a few months: the tumor recurred in one case in spite of surgery, chemotherapy (specific RMS protocol), and radiation therapy; the tumor recurred in one other case after surgery; and one child did not receive any treatment [
127,
139,
141]. The outcome was not known or favorable in other cases. Interestingly, one case exhibited a t(2;8)(q35;q13) translocation. When it was performed, cytogenetic techniques never evidenced any
FOXO1 fusion. One case was associated with Apert syndrome (craniosynostosis, midface hypoplasia, and syndactyly of the hands and feet), a syndrome driven by mutations in
FGFR2. In a recent study of germline predisposition variants in pediatric RMS, the authors found some cancer predisposition gene variants in ERMS occurring before 1 year of age, but not in ARMS in this age group. The exact age and location of the tumors are not discussed in the paper, but the most frequently involved genes have been
NF1 (neurofibromatosis type 1),
TP53 (Li–Fraumeni syndrome),
HRAS (Costello syndrome),
CBL (Noonan syndrome), and
BRCA2 [
142]. It is worth mentioning that some cases of congenital/neonatal ERMS may arise on congenital melanocytic nevi [
141].
4.3. Rhabdomyosarcoma Risk Group Stratification and Treatment
The outcome and treatment of RMS are dependent on risk groups. Of note, the perineal location seems to have prognostic significance as well, with a poorer prognosis in those cases [
126].
The stratification of patients into risk groups at diagnosis is essential. The definition of risk groups is different in Europe and in North America, with different treatment strategies. In Europe, four risk groups (low, standard, high, and very high) have been defined by the European Paediatric Soft Tissue Sarcoma Group (EpSSG), based on the Intergroup Rhabdomyosarcoma Study Group (IRS) post-surgical stage, the age, the tumor size, the histopathological subtype, the site of the primary tumor, and the nodal stage. According to the EpSSG, surgical resection follows induction chemotherapy when any residual mass can be completely excised (R0/R1) without causing a significant organ or functional impairment. For some standard risk cases, radiation therapy may be avoided if the resection is complete (R0). For more details on the management of RMS and the differences between North American and European approaches, the reader is referred to the review by Yechieli et al. [
143].