Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
In immunology, the discovery of regulatory T (Treg) cells was a major breakthrough. Treg cells play a key role in pregnancy maintenance, in the prevention of autoimmune responses, and in the control of all immune responses, including responses to self cells, cancer, infection, and a transplant.
- regulatory T-cell memory
- Treg cells
- immune memory
1. Introduction
Regulatory T cells (Tregs) are a specialized subset of immunosuppressive CD4+ T cells that express lineage-specific transcription factor forkhead box P3 (Foxp3) [11,12]. Treg cells ensure immunological homeostasis by substantially suppressing autoreactive CD4+ T cells that have escaped negative selection in the thymus [13]. Besides, Treg cells act as key negative regulators of inflammation in various pathological conditions, including infections, autoimmune diseases, and cancer [14,15,16,17]. Treg cells drastically increase their suppressive function in response to inflammation. Activated Treg cells raise the levels of immunosuppressive proteins (IL-10 and/or TGF-β) and chemokines, and they undergo polycomb-mediated repression of Foxp3-bound genes; this process may prevent the acquisition of proinflammatory functions [12]. Treg cells that infiltrate wounds express epidermal growth factor receptor (EGFR) and take part in tissue regeneration. A conditional knockout of EGFR in Treg cells delays wound closure and enhances the accumulation of proinflammatory macrophages [18].
2. The Origin of Treg Cells
During infection, under the influence of signals from a Toll-like receptor (TLR) [19] of proinflammatory cytokines [20] or of costimulation through CD40 [21], dendritic cells are activated, an effector response is triggered, and induced Treg cells develop [22]. This notion is supported by the involvement of the same transcription factors in the differentiation of phenotypically similar subpopulations of CD4+ effector T cells and Treg cells, although there are key differences. For example, transcription factor T-bet is required for the differentiation of T helper 1 (Th1) cells and the differentiation of T-bet+ Treg cells (Figure 1). In response to type I interferon (IFN), IFN-γ or interleukin-27 (IL-27), STAT 1 (signal transducer and an activator of transcription 1) are phosphorylated, and T-bet expression is triggered. T-bet in turn induces the expression of IL-12Rβ2 on the cell surface and enhances the sensitivity of Th1 cells, not of Treg cells, to IL-12 [23,24]. IL-12 activates STAT4 resulting in the T-bet upregulation required for full differentiation of Th1 cells. In contrast to Th1 lymphocytes, the activation through T-bet in Treg cells does not elevate IL-12Rβ2 expression and IFN-γ production [23]. Therefore, the sensitivity of Treg cells to IL-12–dependent differentiation does not increase owing to the presence of inhibitory tri-methyl histone marks (H3K27) in the Il12rb2 promoter [23]. Nevertheless, after prolonged stimulation with IL-12, these Treg cells lose their suppressive function and begin to produce a large amount of IFN-γ [25]. In type 1 diabetes mellitus and in multiple sclerosis, an elevated number of Treg cells secreting IFN-γ is associated with disease aggravation. This observation suggests that reprogrammed Treg cells may contribute to autoimmunity pathogenesis [26,27].
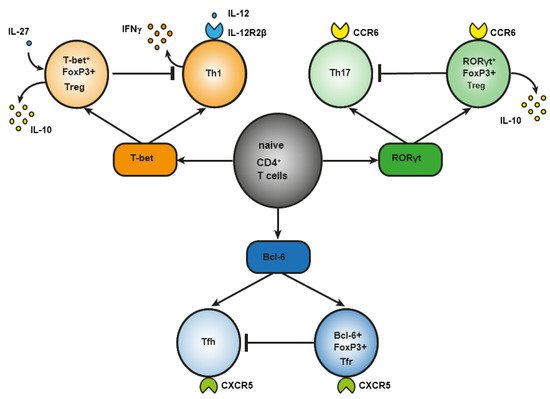
Figure 1. Differences between effector CD4+ T lymphocytes and Treg cells in the role of a master regulator in their differentiation.
Regulatory T (Treg) cells constitute a specific anti-inflammatory lineage of CD4+ T-lymphocyte differentiation determined by X-linked transcription factor Foxp3 [28]. Nonetheless, some activated Treg cells can express transcription factors (T-bet and RORγt) characteristic of effector CD4+ T cells [24,29,30,31]. In a steady state, activated Treg cells temporarily upregulate T-bet for their own homeostasis [31]. Among tumor-infiltrating Tregs, investigators detected T-bet+Foxp3+CD4+ T cells with a higher expression of typical Treg factors, such as ICOS, GITR, CD103, CTLA4, PD-1, and IL-10 as compared to CD4+ T cells, which express either T-bet or Foxp3 [32]. T-bet–deficient Treg cells are characterized by reduced survival and by a failure to inhibit Th1-mediated inflammation after adoptive transfer into Scofy mice [33]. On the other hand, there are several studies indicating that T-bet deficiency does not affect the suppressive function of Tregs [34,35]. In the small-intestine lamina propria, a population of resident RORγt+FoxP3+ Treg cells was found that has an activated phenotype (CD44highCD62Llow) and strongly expresses typical Treg factors ICOS and CTLA-4 and nucleotidases CD39 and CD73 [29]. The presence of RORγt+FoxP3+ Treg cells is crucial, and it reduces the risk of colitis and colorectal cancer [36]. There are fewer RORγt+ Tregs in the gut of patients with food allergy, and commensal-bacteria–mediated protection from food allergy depends on RORγ+ Tregs [37]. Resident RORγ+ Tregs and Th17 lymphocytes reduce the severity of M. tuberculosis–driven lung inflammation in mice [38]. Bcl-6+FoxP3+ Treg cells strongly express coinhibitory protein PD-1. In mice, B cell lymphoma (Bcl) 6 deficiency in Foxp3+ Tregs aggravates experimental Sjögren’s syndrome, but at the same time it enhances immunity to an influenza virus [39]. In breast cancer, Bcl+FoxP3+ Treg cells promote the formation of the B cells that produce IL-10 [40]. Abbreviation: bcl-6, B cell lymphoma (Bcl) 6; CCR, C-C chemokine receptor; CTLA-4, cytotoxic T-lymphocyte-associated protein; CXCR, CXC chemokine receptor; FoxP3, forkhead box; GITR, glucocorticoid-induced tumor necrosis factor-related receptor; ICOS, inducible costimulator; IL, interleukin; PD-1, programmed cell death protein 1; RORγt, RAR-related orphan receptor gamma; Tfh, T follicular helper cells; Tfr, T follicular regulatory cells; Treg cells, regulatory T-cells.
Another example of differences in the role of a master regulator of T-cell differentiation is RAR-related orphan receptor gamma (RORγt). It is known that RORγt is important for the differentiation of Th17 lymphocytes and for the expression of C-C chemokine receptor (CCR) 6 [41]. The signaling initiated by cytokines of the IL-17 family via STAT3 is known to cause the differentiation of Th17 lymphocytes [42]. On the other hand, in the lamina propria of the small intestine in humans and mice, a population of Treg cells has been found that also expresses RORγt [43,44] and CCR6 [45]. The production of such RORγt+ Tregs involves STAT3-dependent transcriptional pathways (similar to those in Th17 lymphocytes), and it requires microbiota antigens for their differentiation [44,46]. Loss of STAT3 leads to downregulation of CCR6 in RORγt+ Tregs, and it impairs their migration to the intestine [47]. In Th17 cells, IL-6 is required for the activation of a factor called STAT3, whereas IL-10 is required for this process in Treg cells [47]. CCR6 may direct the migration of Treg cells to sites of Th17-mediated inflammation, suggesting that these CCR6+ Treg cells may be especially potent suppressors of Th17 responses [48]. Mice that are deficient in RORγt+FoxP3+ Tregs develop a severer and more lethal type of oxazolone-induced colitis, which is a model of ulcerative colitis [36,49,50]. Nevertheless, in the presence of IL-1, IL-23, IL-6, and transforming growth factor beta (TGF-β), naïve CD25+FoxP3+ Treg cells show an upregulation of RORγt—along with downregulation of FoxP3 and a loss of suppressor functions—and they differentiate into Th17 lymphocytes [51].
Bcl-6+ “T follicular regulatory” (Tfr) cells express B-cell–associated CXC chemokine receptor (CXCR)5 and develop in parallel with Bcl-6+ T follicular helper (Tfh) cells, which contribute to humoral immunity [52,53]. Tfr cells also express a Bcl-6 antagonist called Blimp-1, which is not expressed by Tfh cells [54]. Transcription factor NFAT2 is required for CXCR5 expression in Tfr cells but not in Tfh cells [55], thus further supporting the notion that effector T cells and Tregs use different molecular pathways to attain similar phenotypes. The main function of Tfr cells is thought to be the suppression of the germinal cancer reaction and the inhibition of B-cell proliferation and immunoglobulin production [40].
Accordingly, it is obvious that Treg cells are a heterogeneous population. Depending on their origin, Tregs are categorized into two subpopulations: natural (nTregs) and induced Tregs, i.e., those formed in secondary lymphoid organs during an immune response (iTregs) [11,13,56]. In mice, transcription factor Helios was identified as a marker discriminating between thymic Treg cells (Helios+) and peripheral Treg (pTreg) cells (Helios−) [57]. In the present discussion, it is assumed that nTreg cells are (i) Tregs that arise in the thymus during negative selection of CD4+ T lymphocytes (tTregs) [12] and (ii) Tregs originating in the periphery when stimulated by the commensal microbiota (pTregs) [58,59]. According to the literature, >90% of Treg cells of adipose tissue [60], ~70–80% of Treg cells of the intestine in newborn mice, and ~30% of Treg cells in the intestine of adult animals [61] are of thymic origin, as are Treg cells of the skin [62]. There is a hypothesis that such pTreg cells of barrier tissues constitute a major proportion of mTreg cells and are similar in function to tissue-resident memory T cells [59,62,63]. This theory is supported by evidence that pTreg lymphocytes in the gut and in the skin are necessary to suppress immune responses against local commensal bacteria [62,64,65,66].
A number of subpopulations of Treg cells can be distinguished depending on the phenotypic profile, too. First of all, there are CD4+ Treg cells and CD8+ Treg cells [67,68,69,70]. Second, depending on the expression of CD25, FoxP3, CD127, CD39, CD45RA, CTLA4, GITR, Helios, ICOS, PD-1, FasL, and perforin as well as the secretion of cytokines IL-10, IL-35, and TGF-β, Treg cells can be further categorized into subpopulations that have been described previously [71]. A distinctive feature of Treg cells is the expression of the transcription factor FoxP3 [56], which is required for the establishment and the maintenance of their suppressor activity [10]. Loss of Foxp3 expression leads to the development of a lethal multiorgan autoimmune disease in mice and humans [72,73]. On the other hand, there are FoxP3− Treg cells: type 1 regulatory cells, and IL-35-Producing T Cells (iTR35). They do not express FoxP3 but produce the immunosuppressive cytokine IL-10 [74], and iTR35 cells also secrete IL-35 [75].
It is possible that similarly to subpopulations of CD4+ T cells, Treg cells should be categorized not by phenotypic profiles [76,77] but rather by participation in the suppression of certain effector functions. Depending on microenvironmental factors, differentiating CD4+ Treg cells acquire either suppressor or effector potential. This approach may facilitate the search for specific mTreg markers, which are still being discussed [78].
This entry is adapted from the peer-reviewed paper 10.3390/cells11101687
This entry is offline, you can click here to edit this entry!