- copper catalysis
- carbene intermediate
- alkyne functionalization
1. Alkynylation
1.1. Alkynylation Terminated by Protonation
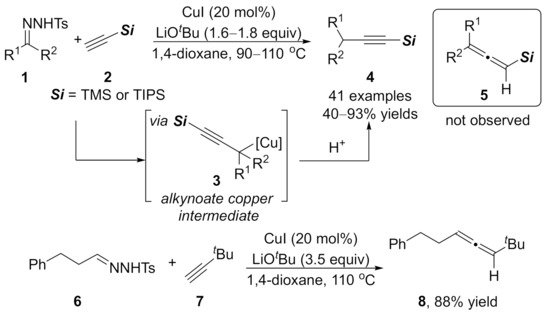
1.2. Alkynylation Terminated by Electrophilic Addition
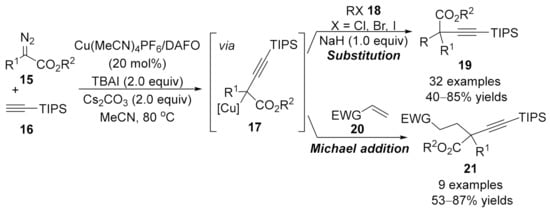
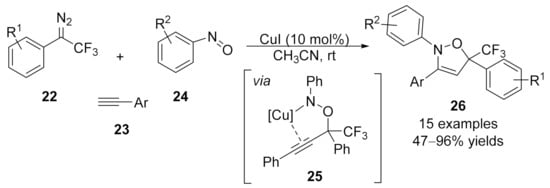
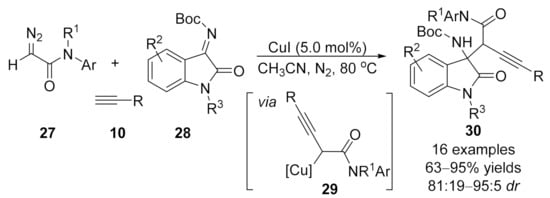
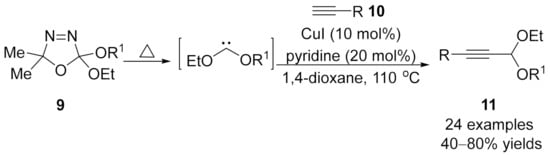
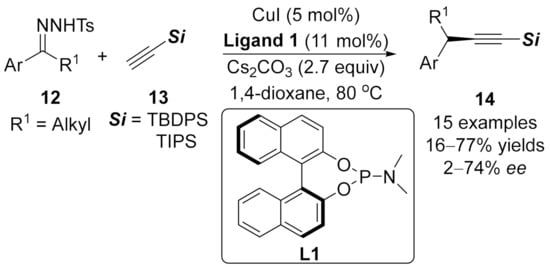
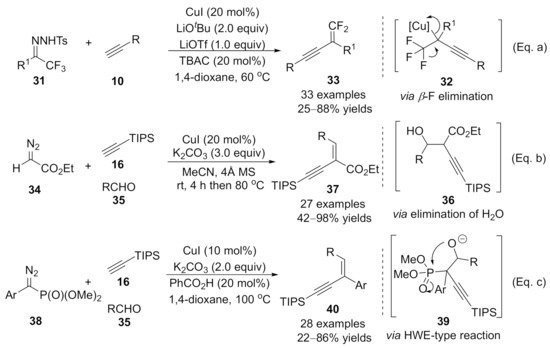
1.3. Alkynylation Terminated by β-Elimination
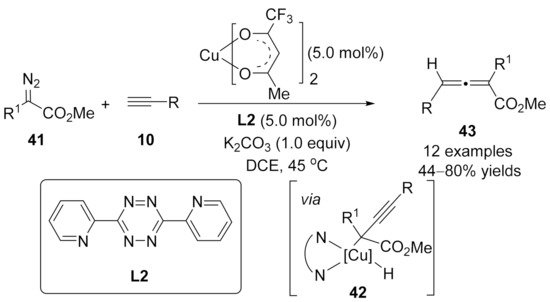
2. Allenylation
2.1. Allenylation Terminated by Protonation
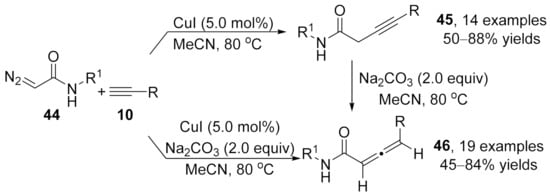
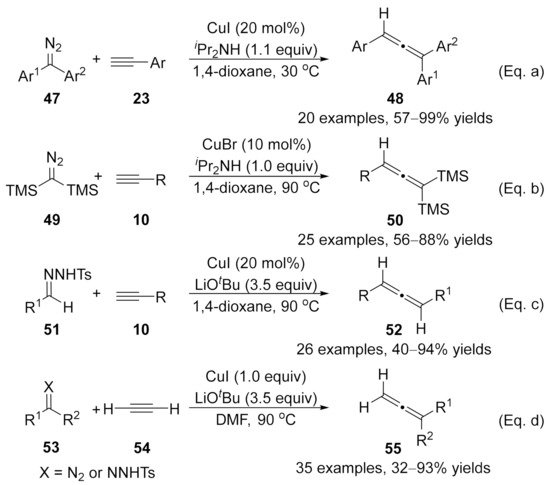
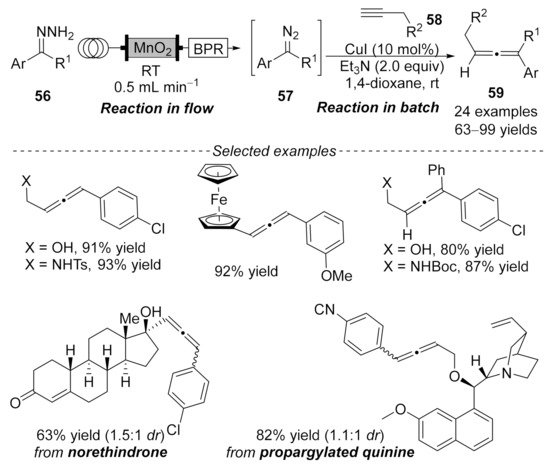
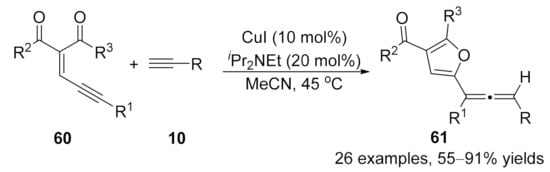
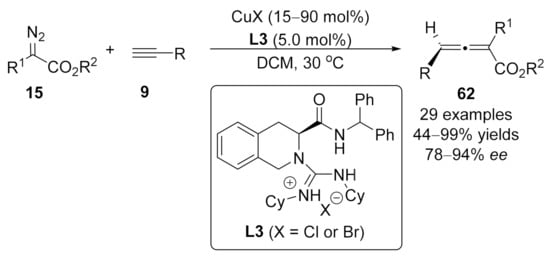
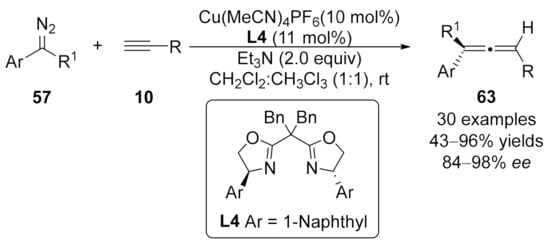
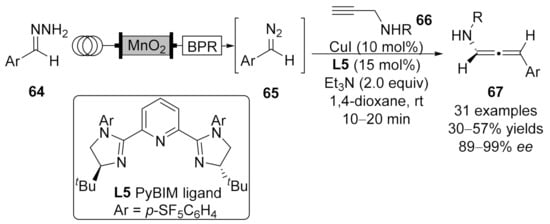
2.2. Allenylation Terminated by Electrophilic Addition
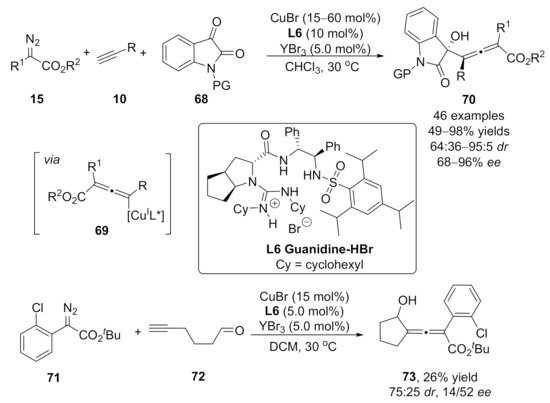
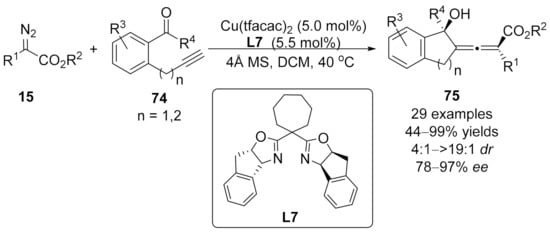
2.3. Allenylation Terminated by Allylation
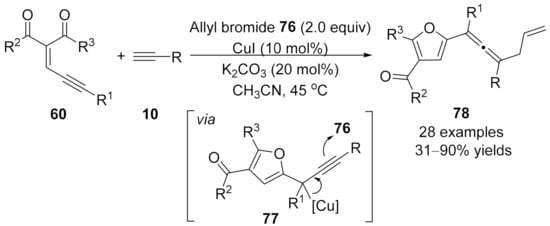
2.2.4 Cascade Transformations Involving Allenylation Process
Nucleophilic addition with allenoic ester or its isomeric compound is generally used synthetic strategy for the expeditious construction of highly functionalized carbocycle or heterocycle structures [24-32]. Thus, a variety of inter- or intramolecular cascade reactions have been developed through different nucleophilic addition processes of the allene derivatives generated from cross-coupling between alkynes and copper carbenes.
In 2011, a one-pot synthesis of phenanthrenes 81 via ligand-free CuBr2-catalyzed coupling reaction/intramolecular cyclization of terminal alkynes 23 with N-tosylhydrazones 79 derived from o-formyl biphenyls was developed by Wang and co-workers [33]. Allene intermediates 85 were initially generated in this cascade reaction through a cross-coupling reaction of N-tosylhydrazones 79 with terminal alkynes 23, followed by a 6π-cyclization and isomerization deliver the phenanthrene products 81 with broad functional group compatibility (Scheme 19).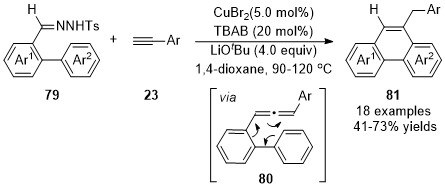
Scheme 19. Copper-catalyzed allenylation followed by 6π-cyclization.
Later in the same year, instead of using o-aryl substituted N-tosylhydrazones, o-hydroxy- or o-amino-substituted N-tosylhydrazones were introduced by the same group as carbene precursors in an analogous cascade transformation, a ligand-free CuBr-catalyzed coupling reaction/intramolecular cyclization sequence, achieving the synthesis of benzofuran or indole derivatives 84 in moderate to excellent yields [34]. The initially formed allene intermediates 83 were trapped through a nucleophilic addition by the embedded o-hydroxy- or o-amino group to afford the cyclized products 84 (Scheme 20).
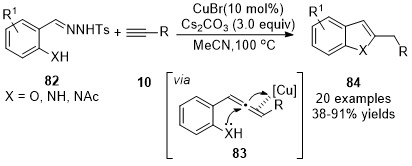
Scheme 20. Copper-catalyzed allenylation followed nucleophilic addition.
In 2011, a similar catalytic strategy was developed by Balakishan’s group. They reported a simple procedure for synthesizing aza- and oxacycles via a copper-catalyzed coupling reaction of functionalized terminal alkynes 85 with diazoesters 86 [35]. Initially, the allene intermediates were formed in the presence of CuI, followed by an intramolecular aza- or oxa-Michael cycloaddition and isomerization to generate the cyclized five- or six-membered products 87 in generally good yields (Scheme 21).
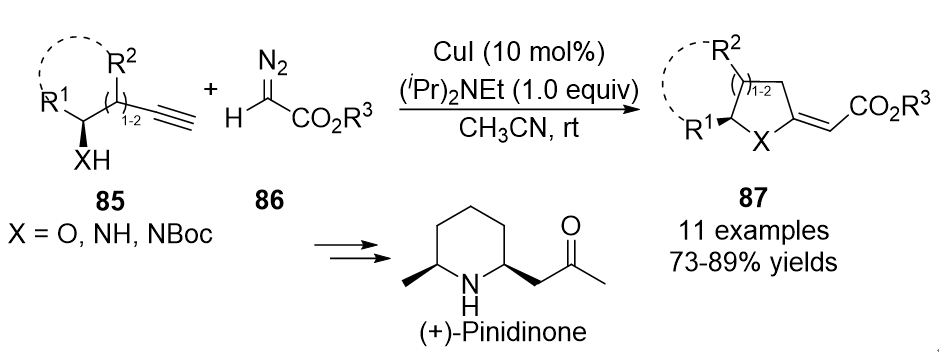
Scheme 21. Copper-catalyzed allenylation followed by cyclization.
In 2015, a stereo-divergent synthesis of five-membered heterocycles was developed by Sun’s group [36]. This work described a copper-catalyzed cross-coupling reaction and annulation cascade reaction of amino alkynes 88 with diazo compounds 15. The proposed reaction mechanism involves trapping in situ formed allene intermediates, yielding 2-methylenes 89 (when PG = Bn) and 2,3-dihydropyrroles 90 (when PG = Ts) in good yields with broad functional group tolerance under mild conditions. Control experimental results show that N-benzyl amino alkynes were more likely to form 2-methylenespyrroles derivatives 89 through 5-exo-dig cycloaddition, while 2,3-dihydropyrroles 90 generated from N-tosylamino alkynes through 5-endo-dig cycloaddition would be more favorable (Scheme 22).
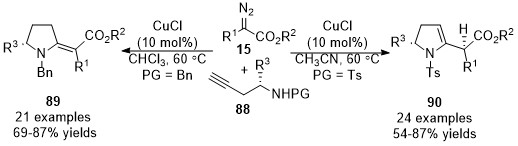
Scheme 22. Copper-catalyzed allenylation followed by divergent annulation.
In 2018, Sun and co-workers expanded the above chemistry to synthesize the four- to six-membered heterocycles with N-substituted prop-2-yn-1-amines 91 and diazoacetates 15 [37]. Generated allenoic species 92 have been proven as the key intermediates for the subsequent diverse annulations under optimized conditions toward functionalized heterocycle in moderate to good yields. Treatment of allenoates 92 with sodium phenolates led to six-membered products 93; silver nitrate and triethylamine yielded five-membered products 94; what’s more, four-membered products 95 were generated under lithium tert-butoxide conditions (Scheme 23).
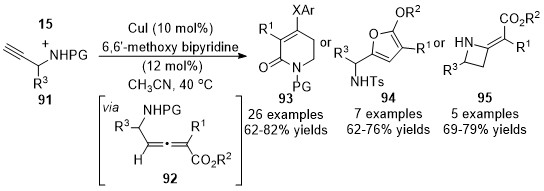
Scheme 23. Divergent synthesis of four- to six-membered heterocycles involving an allenylation process.
In addition to the cyclization through addition with a heteroatom, carbon-based nucleophilic species could also be served as the nucleophile to addition with these allenes, forming the C-C bond instead of the C-X bond [38,39]. In 2015, Kumaraswamy’s group developed a cooper catalyzed cross-coupling reaction/intramolecular Michael addition cascade reaction [40], achieving the formation of indene and dihydronaphthalene derivatives 97 in good yields with broad functional group tolerance (Scheme 24a). Later in 2017, Sun’s group reported an analogous approach toward five- or six-membered carbo-/heterocycles with diazo compounds 15 and alkyne-substituted malonates 98 [41]. In this reaction, the ligand significantly enhanced the reaction yields and inhibited the Conia-ene side reaction. As a result, the polyfunctionalized cyclohexenes, tetrahydropyridines, and dihydropyrans have been prepared in moderate to high yields under mild reaction conditions (Scheme 24b).
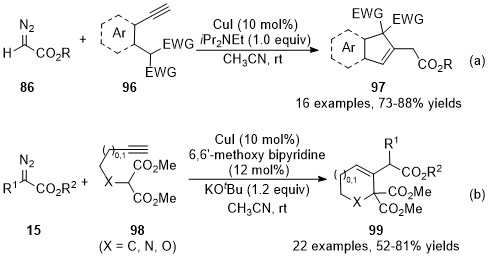
Scheme 24. Copper-catalyzed allenylation followed by Michael addition.
In 2015, a Cu(I)-catalyzed denitrogenative annulation reaction of pyridotriazoles 100 with terminal alkynes 10 was developed by Gevorgyan’s group [42]. Initially, α-pyridyl copper carbenes were generated from pyridotriazoles 100 in the presence of the copper catalyst, followed by a cross-coupling reaction with terminal alkyne to form either propargylic or allenoic intermediates 101, which were terminated by copper-catalyzed cycloisomerization to furnish the indolizines 102 in moderate to excellent yields (Scheme 25).
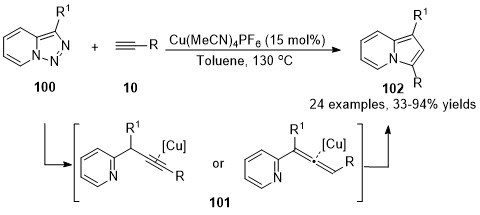
Scheme 25. Copper-catalyzed allenylation followed by cycloisomerization.
In 2018, Wang and co-workers reported a copper-catalyzed geminal difunctionalization reaction of terminal alkynes [43]. The key step in this cascade reaction is trapping the in situ generated allenoic species 105 with a sulfonyl anion to form the carbon-sulfur bond, providing a variety of vinyl sulfones 106 in good yields with excellent stereoselectivities under mild reaction conditions. It was noted that the excellent stereoselectivities might be due to the influence of steric hindrance, and no ligand and additive were required in this transformation (Scheme 26).
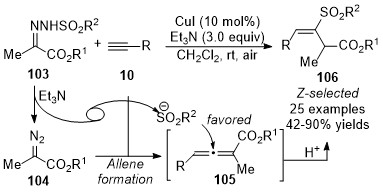
Scheme 26. Copper-catalyzed allenylation followed by sulfonylation.
Recently, Sun and co-workers demonstrated a copper-catalyzed three-component reaction of terminal alkynes with diazo compounds and B2pin2 for the synthesis of trisubstituted alkenylboronates [44]. Copper catalysts played dual roles in these alkynes’ difunctionalizations. Initially, copper catalyzed the cross-coupling to form an allenoic intermediate, followed by a copper-catalyzed stereoselective boration reaction with B2pin2. When diazo compounds 53 were used as carbene precursors, the steric interaction forced the boron group to attack the β-carbon from the opposite side of the γ-phenyl group on the allenoic species 107, leading to the favored (Z)-isomers 108 as major products. Whereas, in the case with N-tosylhydrones 51 as carbene precursors, the addition of Cu-Bpin complex to corresponding allenoic species 109 provided allyl copper intermediate, which was more favored to form a six-membered ring transition state with the association of MeOH, finally furnishing the more thermodynamically stable (E)-products 110 (Scheme 27).
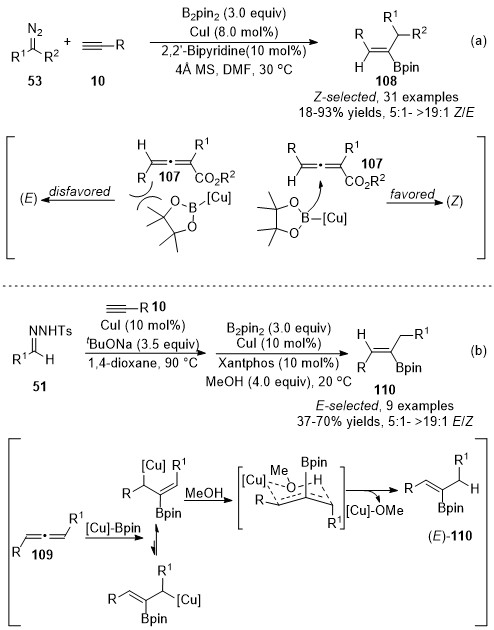
Scheme 27. Copper-catalyzed allenylation followed by boroalkylation.
3.Copper Carbene Intermediate Addition onto C-C Triple Bond
3.1 Cyclopropenation
Cyclopropenation is a well-known reaction of metal carbene intermediate with alkynes. This widely used reaction could be catalyzed by rhodium [45-49], cobalt [50], gold [51], silver [52,53] and many others [54,55,56]. Herein, selected examples related to copper catalysis will be discussed.
In 2010, a new tridentate coordination copper complex, Cu[Ms(CH2SCN)3]BAr'4 (BAr'4 = tetra(3,5-bis(trifluoromethylphenyl)borate), was designed by Miguel and co-workers by using [Cu(OTf)]2•C6H6 and an alkylthiocyanate ligand [150]. This catalyst promoted the cyclopropenation of ethyl diazoacetate 34 (EDA) with a wide range of internal alkynes 111, providing cyclopropenes 112 in moderate yields (Scheme 28). The same cyclopropenation work was achieved by Dias, unique bis(pyrazolyl)borate ligand supported [(CF3)2Bp]Cu(NCMe) catalyst was used, yielding cyclopropene products in moderate to high yields [57].
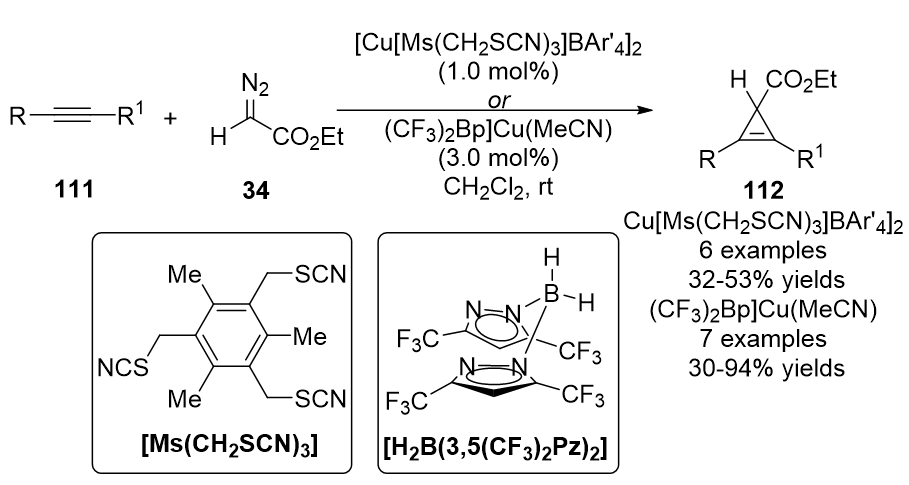
Scheme 28. Copper/alkylthiocyanate complex catalyzed cyclopropenation.
In 2016, a Cu(I)/N-heterocyclic carbene (CuNHC) complex catalyzed cyclopropenation of internal alkynylsilanes 113 with diazoacetate 15 was reported by Coleman’s group [58]. A series of 1,2,3-trisubstituted and 1,2,3,3-tetrasubstituted cyclopropenylsilane compounds 114 were isolated in moderate to good yields (Scheme 29). An interesting regioselective and chemodivergent reaction pathway occurred furnished a tetra-substituted furan through an intramolecular cyclopropene ring-opening transformation in the case of electron-rich diazoacetate.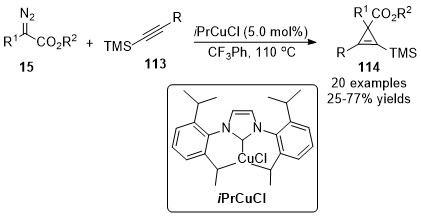
Scheme 29. Copper/N-heterocyclic carbene (CuNHC) complex catalyzed cyclopropenation.
3.2 Cascade Reaction Involving Carbene/Alkyne Metathesis Process
Carbene/alkyne metathesis (CAM) refers to the processes where a metal carbene reacts with an alkyne, generating a new vinyl metal carbene intermediate, which was difficult to access with other carbene precursors [59,60,61]. This in situ generated vinyl metal carbene intermediate could be involved in typical metal carbene reactions, such as [3+2]-cycloaddition [156], cyclopropanation [62,63,64], C-H bond insertion [65-68], and others [69-71]. Herein, we summarized recent works on the copper-mediated cascade transformations involving carbene/alkyne metathesis.
It’s a general protocol for the synthesis of furan derivatives through transition metal-catalyzed formal [3+2] cycloaddition of α-diazocarbonyl compounds with alkynes [72-75]. However, the cases under copper carbenes mediated were limited. In 2014, Wang’s group developed a copper-catalyzed formal [3+2] cycloaddition reaction of terminal alkynes with β-keto α-diazoesters 115 (X = O), offering an operationally simple and applicable method for the synthesis of trisubstituted furans 116 (X = O) with a wide substrate scope (Scheme 30a) [76]. This reaction has also been applied to ethyl (E)-2-diazo-3-(methoxyimino)butanoate 115 (X = NOMe) for the synthesis of 2,3,5-trisubstituted N-methoxypyrroles (X = NOMe). Later in 2016, a Cu(I)-catalyzed cycloaddition of diazoacetates 15 with electron-rich internal aryl alkynes 117 was discovered by Coleman and co-workers [77]. Tetra-substituted furans 118 were generated in moderate isolated yields with high chemoselectivities and regioselectivities (Scheme 30b).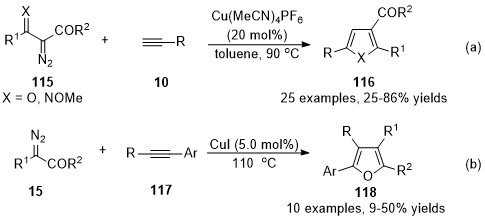
Scheme 30. Copper-catalyzed carbene/alkyne metathesis for the synthesis of furan derivatives.
In 2016, Xu’s group developed a chemo-divergent copper-catalyzed cascade reaction of alkynyl-tethered α-iminodiazoacetates 119, providing polycyclic and multi-substituted pyrroles in high yields with a broad substrate scope [78]. Especially, the tetra-substituted 3-formylpyrroles 124, which were difficult to access by alternate approaches. Mechanistic studies indicated that the α-imino carbene 120 is the key common intermediate in this divergent reaction, which was generated by metal-catalyzed carbene/alkyne metathesis of the alkynyl-tethered diazo compounds 121. When R1, R2 was imbedded with an aromatic ring, polycyclic pyrroles 122 were formed as the major products through a [3+2]-cyclization and aromatization process. Whereas, substrates with a methoxy group on the nitrogen (R2 = OMe), the carbene intermediate underwent an N–O insertion/alkoxy migration/alcoholysis sequence, giving the 3-formylpyrrole products 124 in generally good to excellent yields (Scheme 31).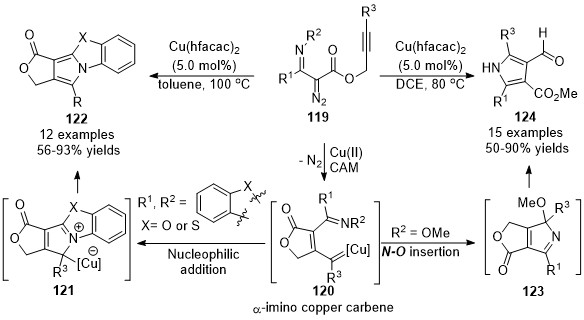
Scheme 31. Copper-catalyzed carbene/alkyne metathesis for the synthesis of pyrroles.
At the same time, Xu and co-workers have also developed a copper-catalyzed carbene/alkyne metathesis cascade reaction with alkyne-tethered diazo compounds 125 [79]. This transformation provided rapid access for the construction of multi-substituted 4-carboxyl quinoline derivatives 127 in high to excellent yields. In this cascade reaction, one C═N and one C═C bond were formed with the assistance of the copper catalyst under mild reaction conditions (Scheme 32a). Later in 2017, Ye’s group reported an analogous protocol by using ynamides 128 as carbene precursor [80]. In this work, a copper carbene was generated in situ through a catalytic oxidation process in the presence of quinoline N-oxide, followed by a CAM process and terminated by carbene reaction with an embedded azide group, providing a wide range of pyrrolo[3,4-c]quinolin-1-ones 130 in good yields. Those works represented practical methods for the dual functionalization of alkynes (Scheme 32b).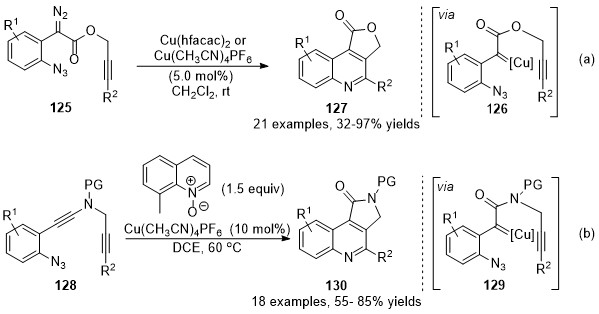
Scheme 32. Copper-catalyzed carbene/alkyne metathesis for the synthesis of quinolines.
In addition to the nucleophilic addition of the in situ formed copper carbene intermediates, electrophilic aromatic substitution or C(sp2)–H bond functionalization is another useful terminating transformation for the direct construction of polycyclic fused frameworks. In 2017, Doyle’s group reported a copper-catalyzed intramolecular cascade reaction of diazo compounds 131, this transformation went through a CAM process followed by a carbene C(sp2)–H bond functionalization cascade, yielding the fused indeno-furanone derivatives 133 in excellent yields under mild reaction conditions (Scheme 33a) [81]. Instead of terminating reaction in C-H functionalization, a selective Buchner insertion reaction occurred as the terminating step in Xu’s work when the ortho-aniline substituted propargyl diazoacetates 134 were employed, selectively affording the dihydrocyclohepta[b]indole derivatives 136 in moderate to high yields (Scheme 33b). Notably, this reaction described a rare example of the Buchner reaction with donor/donor type metal carbene species [82].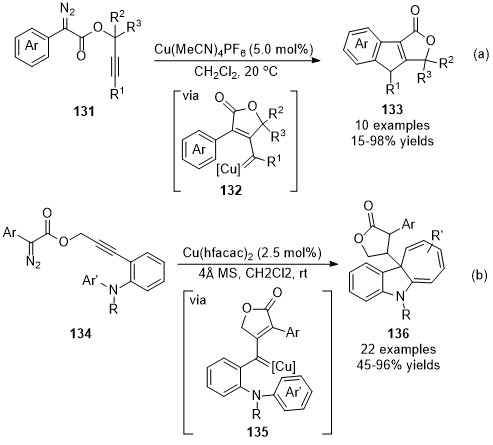
Scheme 33. Copper-catalyzed carbene/alkyne metathesis for the synthesis of tri-cyclic molecules.
In 2018, Xu and co-workers developed an intermolecular copper-catalyzed formal CAM process [83], which underwent a copper promoted [3+2] cycloaddition/dinitrogen exclusion/nucleophilic addition process, providing a direct and effective access to 2H-chromene derivatives 139 in generally good to high yields. Mechanistic studies indicated that the 3H-pyrazole 138 is the key intermediate in this cascade transformation, and this critical intermediate was isolated and confirmed by single-crystal X-ray diffraction and spectroscopy analysis for the first time (Scheme 34).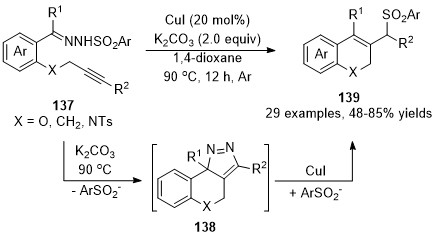
Scheme 34. Copper-catalyzed formal carbene/alkyne metathesis for the synthesis of 2H-chromene derivatives.
Based on a similar protocol, a copper-catalyzed formal [1+2+2]-annulation of alkyne-tethered diazo compounds 140 with pyridines 141 has been reported by Xu’s group recently [84]. In contrast to the previously reported cascade reaction that was terminated the copper carbene intermediate on the carbonic center, a vinylogous addition of vinyl carbene intermediate with pyridine derivatives was occurred in this reaction, followed by an intramolecular annulation to form cycloadducts 146, which underwent a decarboxylative aromatization process to form the desired polycyclic fused indolizine derivatives 147 in good to high yields (Scheme 35, path a), although direct formal [3+2]-cycloaddition via pyridinium ylide pathway could not be ruled out so far (Scheme 35, path b).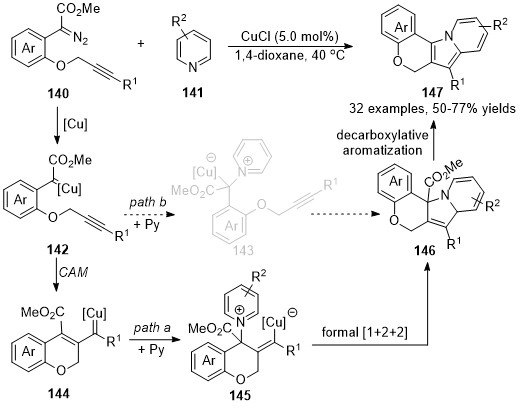
Scheme 35. Copper-catalyzed formal carbene/alkyne metathesis for the synthesis of polycyclic indolizines.
4.4 Conclusions
This review has summarized the recent progress of catalytic alkyne functionalization involving copper carbene intermediates. Copper carbene species derived from different carbene precursors have been introduced to react with alkynes through two distinguished reaction models: cross-coupling reaction of copper carbene intermediates with terminal alkynes and addition of copper carbene intermediates onto the C-C triple bond. The former reaction involves alkynoate or allenoate copper intermediates, followed by protonation, nucleophilic substitution, electrophilic addition, or elimination process, yielding functionalized alkynes and allenes, respectively. The latter version includes cyclopropenation and cascade reaction via carbene/alkyne metathesis process. Although substantial progress has been realized in this field, challenges remain, e.g., the carbene precursors are still mainly limited to the diazo compounds; the catalytic efficiency could be further improved, especially in the asymmetric catalysis; synthetic applications of this chemistry are still under exploration. Therefore, the synthetic potential of this chemistry could be envisioned through the introduction of a variety of readily accessible carbene precursors and with the development of robust copper catalysts/ligands, including novel methodology discovery for the catalytic alkyne functionalization, and expeditious assembly of molecules with structural complexity and diversity for the leading compound development.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27103088