Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Alcohol use disorder (AUD) can be defined as a chronically relapsing disorder characterized by the compulsion to ingest alcohol, the loss of control in limiting alcohol intake despite adverse health, social, and occupational consequences, and the emergence of a negative emotional state that can involve feelings of anxiety, irritability, and dysphoria when access to alcohol is prevented, reflecting a state of motivational withdrawal.
- addiction
- alcohol
- alcohol use disorder
- brain
- neurotransmitter systems
1. Introduction
Alcohol use disorder (AUD) can be defined as a chronically relapsing disorder characterized by the compulsion to ingest alcohol, the loss of control in limiting alcohol intake despite adverse health, social, and occupational consequences, and the emergence of a negative emotional state that can involve feelings of anxiety, irritability, and dysphoria when access to alcohol is prevented, reflecting a state of motivational withdrawal [1]. The 5th Edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V), which was published in 2013, has integrated the previously used terms of alcohol abuse and alcohol dependence into a single condition referred to as alcohol use disorder (AUD). This is measured on a scale of severity ranging from mild to severe, depending on the number of diagnostic criteria met by the patient. There are many factors that influence a person’s susceptibility to alcohol addiction, including age at the onset of consumption, genetic predispositions including family history of AUD, as well as stress and other environmental and socioeconomic factors.
AUD is a serious health condition, and alcohol in general is considered one of the leading preventable causes of death in the United States [3], where 14.4 million adults (ages 18+) and over 400,000 adolescents (ages 12–17) have experienced AUD [4]. Globally, the harmful use of alcohol causes approximately 5.9% of all deaths annually, and 5.1% of the global burden of disease is attributable to alcohol consumption [5].
Chronic exposure to alcohol has profound effects on multiple systems throughout the human body, including the cardiovascular, gastrointestinal, and nervous systems [6]. For the purposes of this review, effects outside of the nervous system are briefly described here. For example, heavy alcohol consumption significantly increases the risk of hypertension, atherosclerosis as well as all forms of stroke [7,8,9,10,11]. Furthermore, alcohol use leads to liver cirrhosis and a range of liver diseases, from liver fibrosis to alcoholic hepatitis [12,13]. Outside of the liver, chronic alcohol consumption can lead to other types of gastrointestinal diseases, including cancers [14,15] as well as acute and chronic pancreatitis [16,17]. Of note, AUD can also alter gut microbiota, which in turn can result in neuroinflammation [18,19].
While alcohol consumption can lead to serious psychosocial dysfunction as well as increased incidence of violence, intimate partner aggression, and suicide [20,21,22,23], prolonged alcohol use and alcohol addiction can also have long-term consequences on the brain and other body systems. Acute alcohol consumption leads to short-term alterations in neurological function primarily due to its actions on inhibitory neurotransmission. Whereas repeated consumption of alcohol over time leads to long-term changes in the functioning of several key neural circuits, causing a compulsion to consume this substance despite adverse consequences as well as the development of a negative emotional state when access to alcohol is restricted [1]. These alterations, among others, are characteristic of AUD and are also commonly associated with addiction to drugs other than alcohol.
2. AUD and the Brain
2.1. Effect of Alcohol on Neurotransmitter Systems
Alcohol (ethanol) has a simple chemical structure that allows it to freely diffuse across the lipid bilayer of cell membranes. As such, alcohol molecules can directly interact with components of the cell membranes, such as receptors and transporters, as well as with several intracellular molecules and structures, thus impacting multiple cellular processes and functions. In particular, alcohol is able to alter synaptic function by impacting multiple neurotransmitter systems, including 5-HT, DA, GABA, Glu, and ACh (Figure 2). The following paragraphs briefly summarize some of the main effects of alcohol on these neurotransmitter systems. A more detailed overview of how alcohol impacts neurotransmission can be found elsewhere [96,97,98,99].
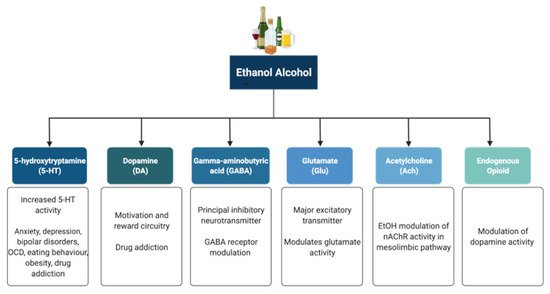
Figure 2. Neurotransmitter systems affected by alcohol (ethanol). Alcohol can interact with multiple neurotransmitter systems in the brain, including the serotonergic (5-HT), dopaminergic (DA), gamma-amynobutyric acid (GABA)-ergic, glutamatergic (Glu), Acetylcholinergic (ACh), and opioid systems, disrupting synaptic transmission and signalling and resulting in the dysregulation of neuronal networks that control reward, motivation, decision making, affect, and the stress response.
5-HT has been implicated in anxiety, depression, bipolar disorders, obsessive compulsive disorders, eating behaviour and obesity, and drug addiction [100,101]. Alcohol has been shown to potentiate the activity of 5-HT3 receptors in murine models using comparable alcohol concentrations to those seen in humans afflicted with AUD [102,103]. Furthermore, genetic variants linked to 5-HT3 receptor sensitivity have been shown to result in an enhanced DAergic reward pathway in humans [104]. In addition, alcohol dependence has been associated with changes in the transcription of the serotonin transporter (5-HTT), which is encoded by the Slc6a4 gene and is responsible for controlling the pattern and magnitude of 5-HT activity [101,105]. Within the Slc6a4 gene, a repeat element of variable length in the 5′ region and a single nucleotide polymorphism (SNP) in the 3′ untranslated region were shown to influence alcohol dependence and severity of drinking as well as response to 5-HT-targeted therapies in AUD patients, respectively [106,107]. Within this scenario, interactive effects of multiple sequence variations at different levels within a specific serotonergic pathway have been proposed to confer greater susceptibility to developing AUD when compared to single variations [108].
DA is known to play a central role in the development of drug addiction, with animal studies suggesting that alcohol administration causes enhanced DAergic neurotransmission within the VTA and a consequent increase in DA levels in the NA [109,110,111]. In AUD, reduced DA receptor sensitivity is thought to decrease motivation for endogenous effectors of the reward circuitry, leading to enhanced compensatory alcohol consumption [112]. Of note, various genetic mutations and polymorphisms that play a role in DAergic neurotransmission have been suggested to contribute to increased vulnerability to alcohol addiction, including the DA receptor D2 Taq1A polymorphism [113,114,115], the DA transporter gene Slc6a3 polymorphism [113,116], and the missense mutation within the catechol-O-methyltransferase (Comt) gene [112,117,118,119]. However, further research is still required to completely elucidate the relationships among genetic factors, DAergic neurotransmission, and the development of AUD.
The endogenous opioid system has important implications for addiction, including modulation of DA release in the NA and of DAergic neurotransmission within the mesolimbic pathway [120]. Polymorphisms of the Oprm1 gene, which encodes the µ-opioid receptor, have been studied in relation to alcohol addiction with mixed results [121,122,123,124,125,126]. Additionally, both the delta and kappa opioid receptors have also been implicated in alcohol addiction [127,128]. Indeed, single nucleotide polymorphisms of Orpk1 and Orpd1 genes may influence behavioural responses to naltrexone [127].
ACh is a neurotransmitter with a wide range of functions both within and outside the central nervous system. SNPs within the cholinergic receptor muscarinic-2 (CHRM2) gene have been associated with predisposition to alcohol and drug dependence and with the development of affective disorders, including major depressive disorder [129,130]. SNPs within the cholinergic receptor nicotinic alpha-5 subunit (CHRNA5) gene have also been associated with alcohol dependence [131].
The eCB system function is also affected by alcohol both acutely and chronically [132], and this system likely plays a complex role in addiction and withdrawal. Acutely, alcohol decreases levels of the eCBs Anandamide (AEA) and 2-arachidonoylglycerol (2-AG) in hippocampal, amygdala, PFC, and cerebellar tissue [133,134,135]. Long-term exposure to alcohol has been documented to reduce both the binding to and expression of the cannabinoid receptor type a (CB1) in the brain [136,137,138,139]. In some cases, these effects can be transient and are not evident after a period of abstinence from alcohol [136,137]. Further research is required in this area in order to better understand how the eCB system is affected by alcohol, as this system has the capacity to influence other neurotransmitter systems responsible for addiction in the brain.
GABA is the principal inhibitory neurotransmitter in the adult human central nervous system. Studies have shown that alcohol allosterically modulates GABAA receptors, and this mechanism may contribute to tolerance, dependence, and withdrawal in AUD [140,141,142]. The sensitivity of GABAA receptors to alcohol has been suggested to be regulated by phosphorylation of the gamma-2 subunit by protein kinase C (PKC) [143,144]. Disruption of PKCɛ, in particular, appears to disrupt voluntary drinking behaviour in mouse models [145,146]. Alcohol has been shown to enhance DAergic neuronal firing rate via decreased firing frequency of GABAergic units within the VTA and NA, thereby reinforcing the effects of alcohol within the pathways involved in reward [147]. In addition, other studies have shown that alcohol increases GABAergic neurotransmission in the cerebellum, hippocampus, and thalamus [148,149,150]. Furthermore, some studies have suggested a potential link between the presence of specific haplotypes within the GABRA2 gene responsible for encoding the α2 subunit of the GABA receptor and susceptibility to developing AUD [151,152,153,154].
Glu is the major excitatory neurotransmitter in the human brain. Acute alcohol exposure generally inhibits Glu neurotransmission, whereas chronic exposure and acute withdrawal have the opposite effect [155]. Alcohol likely affects Glu neurotransmission by altering the function of both metabotropic (mGluRs) and ionotropic (iGluRs) Glu receptors. Upregulation of the metabotropic glutamate receptor 5 (mGluR5)-Homer2-phosphoinositide 3-kinase (PI3K) signalling pathway by binge drinking has been hypothesized to predispose toward a high binge-like alcohol-drinking phenotype [156]. In addition, abnormal hyperactivation of Ras-extracellular signal-regulated kinase (ERK) downstream of mGluR5 results in a hyper-glutamatergic state and has been thought to be a key factor in behaviours associated with addiction [157]. Alcohol drinking was also shown to attenuate the function of D2 DA autoreceptors and group II mGluRs within the posterior VTA [158]. On the other hand, alcohol has inhibitory effects on iGluRs, being capable of inhibiting NMDA receptors [159,160,161]. However, chronic alcohol exposure was shown to increase postsynaptic NMDA receptor function in the rat basolateral amygdala [162]. Of note, the relationship between both the NR2A and NR3A NMDA receptor subunits and susceptibility to addiction has also been investigated, with studies showing a role for these subunits in alcohol dependence and acute NMDA receptor sensitivity to alcohol [163,164]. Variations in the NMDA-dependent α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor trafficking cascade controlling Glu-related excitatory neurotransmission have also been associated with alcohol dependence [165]. Alcohol has also been shown to reduce NMDA receptor expression and function in the NA and to cause deficits in NMDA receptor-dependent long-term depression (LTD) in this brain region after protracted withdrawal [166]. In addition, chronic intermittent ethanol exposure (CIEE) was shown to affect kainate receptors and result in postsynaptic increases in Glu neurotransmission [167] while also increasing the amplitude and frequency of AMPA-receptor-mediated spontaneous excitatory postsynaptic currents in the rat basolateral amygdala [162]. Of note, microinjection of the AMPA-receptor antagonist 6,7-dinitroquinoxaline-2,3-dione (DNQX) was capable of attenuating withdrawal-related anxiety-like behaviours, suggesting that increased Glu function may contribute to anxiety during withdrawal from chronic alcohol exposure [162]. A more detailed review of the effects of alcohol on Glu reward circuitry can be found elsewhere [168].
Acute and chronic alcohol exposure has also been shown to affect synaptic plasticity, therefore influencing the efficacy of synaptic transmission at synapses. As explained above, alcohol can directly impact the major excitatory (i.e., glutamatergic) and inhibitory (i.e., GABAergic) neurotransmitter systems within the adult central nervous system, thus effectively contributing to changes in both long-term potentiation (LTP) and LTD, and influencing learning and memory processes [169,170]. Of note, pre-natal alcohol exposure has also been shown to have profound effects on hippocampal synaptic plasticity during development [171].
2.2. Effects of Alcohol on Other Synaptic Targets
Alcohol has been shown to interact both directly and indirectly with additional synaptic and intracellular signalling targets within the brain, and this topic has been reviewed elsewhere [172]. In this section, we will present a brief summary of the main effects of alcohol on some of the synaptic and molecular targets within the brain and how these can affect synaptic activity.
Small (SK) and large conductance (BK) Ca2+ and voltage-gated K+ channels have been implicated in alcohol tolerance and adaptive plasticity. Chronic alcohol exposure has been shown to reduce SK channel function in VTA DAergic and CA1 pyramidal neurons and disrupt the SK-channel-NMDA receptor feedback loop, contributing to alcohol-associated adaptive plasticity of glutamatergic synapses [173,174]. Chronic alcohol exposure also leads to enhanced intrinsic excitability and glutamatergic synaptic signalling in lateral orbitofrontal cortical neurons, a mechanism that may contribute to the impairment of behaviours associated with the orbitofrontal cortex in AUD [175], such as anxiety, impulsivity, and aggression [176]. Alcohol has also been shown to interact with BK channels; however, factors such as the level of the activating ligand (intracellular Ca2+), BK subunit composition, post-translational modifications, channel lipid microenvironment, and type of alcohol exposure determine whether or not potentiation or reduction in BK currents occur following alcohol exposure [177]. Alcohol has also been shown to activate G-protein-gated inwardly rectifying potassium (GIRK) channels [178], thereby regulating neuronal excitability and influencing the development of alcohol addiction [179].
In addition to influencing synaptic channels and receptors, there is some evidence that long-term exposure to alcohol may influence synapse structures. Binge alcohol exposure alters scaffolding proteins associated with excitatory synapses [180]. Notably, the morphology of synapses has been shown to be disrupted, and the sizes of dendritic spines are reduced by chronic alcohol exposure in utero, during adolescence, and adulthood in rodent models [181,182,183,184,185,186].
Noteworthy, chronic alcohol use has also been linked to changes in multiple intracellular signalling pathways that can affect synaptic function directly or indirectly. These include alterations in adenosine signalling [187,188], as well as changes in PKC and adenylate cyclase activity [189,190,191].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10051192
This entry is offline, you can click here to edit this entry!