MCPIP1 (also known as regnase-1) is encoded by the ZC3H12A gene and is composed of 599 amino acids that encode a 66-kDa protein. MCPIP1 is a potent anti-inflammatory protein, and plays many roles within the regulation of the immune response.
- MCPIP1
- inflammation
- immune response
- chronic inflammation
- interleukins
- RNase
1. Introduction
MCPIP1 is a multifaceted anti-inflammatory protein that plays a critical downregulatory role in the immune inflammatory response through at least two independent mechanisms: destabilising the mRNA transcripts of many cytokines and inhibiting lipopolysaccharide (LPS)- and IL-1β-induced NF-κB signalling [1].
MCPIP1 possesses a well-documented ribonuclease ability, as demonstrated by its targeting of many cytokine mRNAs for direct degradation; specifically, previous studies have shown that MCPIP1 degrades IL-1β, IL-6, IL-8, IL-12p40, and IL-17 mRNA transcripts (Table 1) [2][3][4][5][6][7][8].
Table 1. mRNA of cytokines and chemokines degraded by MCPIP1 in various types of cells.
|
mRNA |
Cells |
References |
|
IL12p40 |
Macrophages |
[3] |
|
IL6 |
Macrophages, cardiomyocytes |
|
|
IL1β |
Macrophages |
[2] |
|
IL17RA, IL17RC |
Lymphocytes, fibroblasts |
[5] |
|
IL8 |
Epithelial cells |
[4] |
|
IL2 |
T cells |
[7] |
|
Cxcl1, Cxcl2, Cxcl3 |
- |
[8] |
The regulation of mRNA transcripts is one of the primary mechanisms through which protein levels are controlled: these molecules can be protected or destroyed to alter the amount of protein being translated under specific circumstances.
Across all organisms, mRNA destabilisation and decay can be performed via a variety of pathways, such as the targeting of conserved AU-rich elements (AREs) and stem-loop structures (SLs) or nonsense-mediated decay (NMD), which prevents the translation of mRNAs. Mino and colleagues discovered that MCPIP1 degrades IL-6 mRNA via the SL structure in the 3′-UTR region, and IL-6 mRNA molecules lacking this sequence were not degraded [8]. More recently, Wilamowski et al. observed that IL-6 is degraded by MCPIP1 in a progressive manner: after SL is cleaved, multiple shorter single-stranded RNA (ssRNA) molecules are generated, and these molecules are then further degraded by MCPIP1. Interestingly, these researchers also found that a 6-nt RNA molecule was bound but not degraded, possibly because it is too short to reach the catalytic site [9].
The critical RNase capability of MCPIP1 is due to its PIN domain (Figure 1) [9]. PIN domains, which are a common motif found in both prokaryotic and eukaryotic nucleases, are primarily responsible for binding to and degrading RNA molecules and also play roles in the bacterial stress response and pathogenesis [10]. The importance of the PIN domain was demonstrated by Matsushita and colleagues, who used site-directed mutagenesis to alter one amino acid (D141N) and found that this alteration completely abolished the RNase function [3].
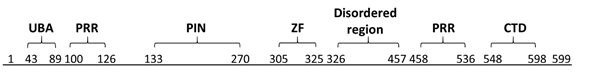
Figure 1. Domains of MCPIP1. Ubiquitin-associated domain (UBA) 43–89; proline-rich region (PRR) 100–126 and 458–536; PilT N-terminus nuclease domain (PIN) 133–270; zinc-finger motif (ZF) 305–325; disordered region 326–457; and C-terminal conserved domain (CTD) 545–598. Based on Wilamowski et al., 2018 [9].
Within the PIN domain, four aspartate residues act in coordination with a single magnesium ion to assemble a catalytic cleft, which constitutes the active site of MCPIP1. This positively charged loop sequence might be responsible for binding to RNA by specifically attracting the negatively charged phosphate groups of oligonucleotide backbones [11].
The USP10-dependent deubiquitination of NEMO and TRAF6 proteins is another strategy through which MCPIP1 can regulate inflammation and the immune response [12]. It results in the negative regulation of the transcription factors c-Jun N-terminal kinase (JNK) and NF-κB [13]. JNK and NF-κB signalling mediate many cellular responses, including infections, inflammation, and apoptosis, through the transcriptional activation of several cytokine genes (Figure 2) [14].
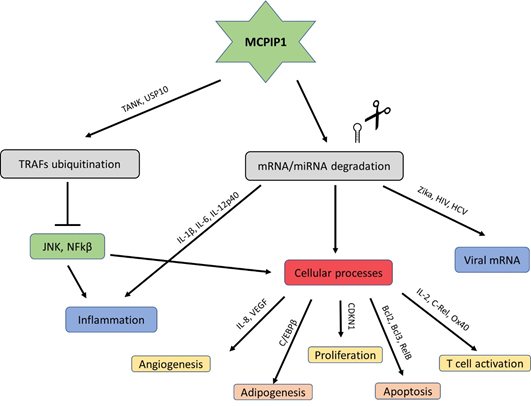
Figure 2. Schematic modes of action of MCPIP1.
MCPIP1 also possesses a CCCH zinc finger domain close to the C-terminal region of the PIN domain (Figure 2). Zinc finger domains are commonly regarded as DNA-binding, but CCCH-type zinc fingers, among others, are involved in binding to RNA molecules and regulating their metabolism [15]. CCCH zinc finger proteins are particularly associated with immune responses and play roles in antiviral innate immunity, the production of cytokines, immune cell activation, immune homeostasis, and the regulation of cell differentiation and cancer cell growth. For example, Roqiun-1, another CCCH-type zinc finger protein, causes a lupus-like autoimmune disease in mice when mutated [16].
MCPIP1 appears to undergo homooligomerisation during interaction with RNA substrates (Figure 3). Size-exclusion chromatography revealed that a dimeric form of MCPIP1 appears to be the most common under native conditions, although tetrameric and monomeric fractions might also be present [9]. This homooligomerisation occurs through the proline-rich C-terminal domain [17]. Mutations that prevent oligomerisation also abolish RNase activity, which indicates that oligomerisation is crucial for MCPIP1 enzyme function [18]. Other RNases and RNase domains also function in an oligomerised state; for example, RNase A oligomerises via a proline-dependent arm exchange mechanism [19].
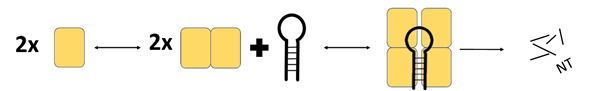
Figure 3. Schematic presentation of the ternary complex model of MCPIP1. Based on the size exclusion chromatography results Wilamowski et al. proposed a sequential binding model: oligo + MCPIP1dimer + MCPIP1dimer ⇄ oligo-MCPIP1dimer + MCPIP1dimer ⇄ oligo-MCPIP1tetramer (MCPIP1 marked as a yelow box). MCPIP1 degrades RNA molecules as a tetramer [9].
The C-terminal region appears to be crucial for another function of MCPIP1: MCPIP1 plays a broad role in suppressing microRNA (miRNA) activity and biogenesis [17]. miRNAs are small non-coding RNA molecules that perform RNA silencing and post-transcriptional regulation of gene expression. MCPIP1 RNase blocks Dicer processing and prevents pre-miRNAs from developing into mature miRNAs. MCPIP1 might recognize and degrade pre-miRNAs in a similar way to mRNAs by targeting terminal loop structures and cleaving RNA with the PIN domain [17]. Both human and viral pre-miRNAs are cleaved by MCPIP1. However, some viral miRNAs might be able to resist this effect and repress MCPIP1 expression, as evidenced by Happel and colleagues, who studied the relationship between MCPIP1 and miRNAs in Kaposi’s sarcoma-associated herpesvirus (KSHV) [20].
2. Broad Roles of MCPIP1 in the Immune System
The crucial role that MCPIP1 plays in the regulation of inflammation can be clearly demonstrated by knockout mice. ZC3H12A-/- mice exhibit a severely altered phenotype characterized by drastic immune system dysregulation: these mice suffer growth retardation and spontaneous death usually within 12 weeks [3]. In these mice, immune cells are overactivated and hyperinvading, resulting in splenomegaly, lymphadenopathy, anaemia, and hyperimmunoglobulinemia. These deviations from normal physiology do not appear until after birth; newborn ZC3H12A-/- mice do not exhibit many obvious differences from their wild-type littermates. After approximately 3 weeks, however, the spleen sizes are notably larger, and leukocyte infiltration into the interstitial spaces of the lungs can be observed. In addition, proinflammatory cytokines, such as IL-6 and IL-1β, exhibit markedly higher expression in MCPIP1-deficient mice.
Interestingly, treatment with antibiotics improves the lifespan and ameliorates hyperinflammatory syndrome in knockout mice [21]. This finding might be explained by host-microbiota interactions: it has been hypothesized that commensal microbiota in mucosal surfaces and the digestive system can elicit TLR signalling and thereby contribute to the development of some autoimmune diseases such as IBD [22]. Therefore, this finding might indicate another function of MCPIP1 within the immune system: attenuation of baseline TLR signalling by resident microorganisms.
The role of MCPIP1 as a broad negative regulator of inflammation is also evidenced by the stimuli that can induce its expression. ZC3H12A expression is induced by a number of proinflammatory factors, although the strongest inducers might depend on the cell type. For example, in human hepatoma HepG2 cells, IL-1β is an effective stimulant, whereas in promonocytic U937 cells, TNF is the strongest inducer of MCPIP1 [2]. The expression of the ZC3H12A gene is also rapidly induced by many factors that are present during infectious attack by pathogens. MCPIP1 expression is increased during viral, bacterial, and fungal infections [23]. LPS, the main outer membrane component of gram-negative bacteria, and the mycobacterium tuberculosis 38-kDa antigen both trigger TLR signalling and subsequently increase MCPIP1 expression [24][25].
However, MCPIP1 appears to exert an overall relaxing effect on immune cell activation. Conditional T-cell MCPIP1-knockout mice exhibit greatly increased rates of T-cell activation, which leads to the conclusion that MCPIP1 plays a role in suppressing the activation of immune cells. MCPIP1 degrades the mRNAs of the genes c-Rel, Ox40, and IL-2, which are responsible for the activation of T cells [26]. MCPIP1 works cooperatively with Roquin to suppress the differentiation of proinflammatory T helper 17 (Th17) cells [27]. Moreover, MCPIP1 negatively regulates group 2 innate lymphoid cells (ILC2s) functions, which are a critical innate source of type 2 cytokines in allergic inflammation. Matsushita et al. discovered that IκB kinase (IKK) complex–mediated MCPIP1 degradation is essential for IL-33– and IL-25–induced ILC2 activation [28].
Despite this effect of limiting immune cell activation, MCPIP1 might play a role in defending the host from foreign nucleic acids such as viruses. Qian and colleagues demonstrated that MCPIP1 can distinguish between mRNAs from exogenously transfected plasmids and those from the host genome and selectively degrade foreign transcripts. Additionally, these researchers showed that the induction of MCPIP1 can restrict Zika virus infection and thereby significantly decrease the viral RNA levels [29][30][31][32]. MCPIP1 also potently inhibits other viral infections, including HIV-1 and Hepatitis C Virus (HCV) infection [21][23].
2.1. MCPIP1 Regulation
The undisputed main role of MCPIP1 is to suppress the inflammatory immune response, which is crucial for preventing a state of chronic inflammation. However, the strong activation of MCPIP1 during pathogen infection would create a favourable environment for the pathogen to invade and replicate. Therefore, pathways need to be in place for the regulation of MCPIP1 and prevent its interference in the immune system’s fight against pathogens. Uehata and colleagues discovered that T-cell receptor activation triggers MCPIP1 degradation by the protease Malt1 [26]. Similarly, the IKK complex ubiquitinates and degrades MCPIP1 after stimulation via TLRs or IL-1β [33].
Another mode of MCPIP1 regulation is the self-degradation of its own transcript [3]. This behaviour establishes a feedback loop to ensure that the immune response is never significantly handicapped by high levels of MCPIP1 expression. Similar to the mechanism through which MCPIP1 degrades many mRNA transcripts, it is thought that this self-regulatory activity is also stimulated by the targeted nucleolytic cleavage of the stem-loop structure contained within the 3′-UTR of MCPIP1 mRNA; luciferase assays with truncated 3′-UTR sequences provide evidence to support this finding [33]. However, another study found that MCPIP1 mRNA without this 3′-UTR sequence is also degraded by MCPIP1 protein [30]. Clearly, further studies are needed to precisely identify the mechanism through which this feedback loop is established.
2.2. Roles of MCPIP1 in the Regulation of Cellular and Bodily Processes
MCPIP1 does not exclusively play important roles in immune function. This factor is expressed in many different cell types and tissues throughout the body. Moreover, MCPIP1 is most highly expressed in leukocytes but is also present in the heart, placenta, spleen, liver, kidney, and lung [3]. This RNase is also involved in many different processes and has subsequently been implicated in many diseases. Many of these involvements are caused indirectly by molecules that regulate or are regulated by MCPIP1. For example, MCP-1, IL-1β, IL-6, and TNFα are involved in different diseases, including rheumatoid arthritis [34], scleroderma [35], COPD [36], diabetes [37], obesity [38], and cancer [39], among many others.
A growing body of evidence shows that MCPIP1 regulates the differentiation and proliferation of many different cell types, and this effect might be related to the MCPIP1-induced increase in reactive oxygen species (ROS), which leads to endoplasmic reticulum stress. For example, Wang and colleagues demonstrated that the forced expression of MCPIP1 induces monocytes into osteoclast precursors and that this effect is accompanied by increased ROS production via the MCPIP1-mediated upregulation of p47PHOX [40]. Similarly, the blockage of MCPIP1 translation with small interfering RNA (siRNA) can result in less ROS production after cholesterol treatment and thereby lower DNA damage [41]. In contrast, MCPIP1 overexpression suppresses the formation of stress granules (SGs) when cells are exposed to arsenite-induced oxidative stress [42]. Although whether MCPIP1 increases or decreases cellular stress levels is currently unclear, the links to cell differentiation are abundant: neural progenitor differentiation to glial cells [43], angiogenic tube formation [44], and adipogenesis induction in 3T3-L1 preadipocytes [45] are all processes regulated by MCPIP1.
2.3. Adipogenesis
In fact, adipogenesis is one of the most well-studied aspects of the non-immune functions of MCPIP1 (Figure 4). Adipogenesis is regulated by a set of transcription factors, including CCAAT/enhancer-binding protein (C/EBP) β, C/EBPδ, C/EBPα and peroxisome proliferator-activated receptor γ (PPARγ) [46]. Although Younce et al. (2009) observed increased adipogenesis after MCPIP1 overexpression [45], later studies observed an opposite effect of MCPIP1 on adipogenesis. In 2014, Lipert and colleagues demonstrated that MCPIP1 impairs adipogenesis: the overexpression of MCPIP1 decreases the C/EBPβ and PPARγ mRNA levels, whereas the silencing of MCPIP1 increases the expression of these mRNAs [47]. Later studies showed that the MCPIP1 level is decreased in the adipose tissue of obese subjects and that MCPIP1 downregulates genes encoding proteins involved in carbohydrate and lipid metabolism while upregulating genes involved in cellular assembly and movement [48].
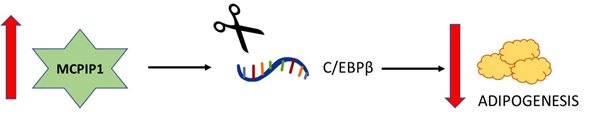
Figure 4. Schematic relationship between the level of MCPIP1 and adipogenesis. A. The overexpression of MCPIP1 decreases the C/EBPβ mRNA level and impairs adipogenesis.
2.4. Angiogenesis
MCPIP1 has also been linked to angiogenesis. Prior to the discovery of MCPIP1, its inducer MCP-1 was associated with angiogenesis: the direct application of MCP-1 to culture media of human aortic endothelial cells (HAECs) results in upregulated expression of hypoxia-inducible factor 1α (HIF-1α) and vascular endothelial growth factor-A165 (VEGF-A165) [49]. Later studies have shown that these proangiogenic effects are instead most likely caused by MCPIP1 rather than MCP-1. The siRNA silencing of MCPIP1 prevents the occurrence of any angiogenic response; the pro-angiogenic cadherin genes Cdh12 and Cdh19 are upregulated by MCPIP1 [44]. The angiogenic capacity of MCPIP1, along with its ability to enhance the cardiac differentiation of mesenchymal stem cells (MSCs), might make MCPIP1 a therapeutic target in the myocardial repair and regeneration of ischaemic tissues [50]. However, more recent studies performed using the clear cell renal cell carcinoma (ccRCC) cell line Caki-1 (metastatic) showed the anti-angiogenic effect of MCPIP1. Caki-1 cells overexpressing MCPIP1 exhibited decreased levels of HIFs, glucose transporter 1 (GLUT1), VEGFA, and IL-6 [51].
This entry is adapted from the peer-reviewed paper 10.3390/ijms21197183
References
- Jolanta Jura; Lukasz Skalniak; Aleksander Koj; Monocyte chemotactic protein-1-induced protein-1 (MCPIP1) is a novel multifunctional modulator of inflammatory reactions. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2012, 1823, 1905-1913, 10.1016/j.bbamcr.2012.06.029.
- Danuta Mizgalska; Paulina Węgrzyn; Krzysztof Murzyn; Aneta Kasza; Aleksander Koj; Jacek Jura; Barbara Jarząb; Jolanta Jura; Interleukin-1-inducible MCPIP protein has structural and functional properties of RNase and participates in degradation of IL-1β mRNA. FEBS Journal 2009, 276, 7386-7399, 10.1111/j.1742-4658.2009.07452.x.
- Kazufumi Matsushita; Osamu Takeuchi; Daron M. Standley; Yutaro Kumagai; Tatsukata Kawagoe; Tohru Miyake; Takashi Satoh; Hiroki Kato; Tohru Tsujimura; Haruki Nakamura; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185-1190, 10.1038/nature07924.
- Ewelina Dobosz; Mateusz Wilamowski; Maciej Lech; Beata Bugara; Jolanta Jura; Jan Potempa; Joanna Koziel; MCPIP-1, Alias Regnase-1, Controls Epithelial Inflammation by Posttranscriptional Regulation of IL-8 Production. Journal of Innate Immunity 2016, 8, 564-578, 10.1159/000448038.
- Abhishek V. Garg; Nilesh Amatya; Kong Chen; J. Agustin Cruz; Prerna Grover; Natasha Whibley; Heather R. Conti; Gerard Hernandez Mir; Tatiana D Sirakova; Erin C. Childs; et al. MCPIP1 Endoribonuclease Activity Negatively Regulates Interleukin-17-Mediated Signaling and Inflammation. Immunity 2015, 43, 475-487, 10.1016/j.immuni.2015.07.021.
- Shigemiki Omiya; Yosuke Omori; Manabu Taneike; Tomokazu Murakawa; Jumpei Ito; Yohei Tanada; Kazuhiko Nishida; Osamu Yamaguchi; Takashi Satoh; Ajay M. Shah; et al. Cytokine mRNA Degradation in Cardiomyocytes Restrains Sterile Inflammation in Pressure-Overloaded Hearts. Circulation 2020, 141, 667-677, 10.1161/circulationaha.119.044582.
- Min Li; Wenqiang Cao; Haifeng Liu; Wei Zhang; Xia Liu; Zhijian Cai; Jing Guo; Xuelian Wang; Zhaoyuan Hui; Hang Zhang; et al. MCPIP1 Down-Regulates IL-2 Expression through an ARE-Independent Pathway. PLoS ONE 2012, 7, e49841, 10.1371/journal.pone.0049841.
- Takashi Mino; Yasuhiro Murakawa; Akira Fukao; Alexis Vandenbon; Hans-Hermann Wessels; Daisuke Ori; Takuya Uehata; Sarang Tartey; Shizuo Akira; Yutaka Suzuki; et al. Regnase-1 and Roquin Regulate a Common Element in Inflammatory mRNAs by Spatiotemporally Distinct Mechanisms. Cell 2015, 161, 1058-1073, 10.1016/j.cell.2015.04.029.
- Mateusz Wilamowski; Andrzej Gorecki; Marta Dziedzicka-Wasylewska; Jolanta Jura; Substrate specificity of human MCPIP1 endoribonuclease. Scientific Reports 2018, 8, 1-14, 10.1038/s41598-018-25765-2.
- M. Senissar; M. C. Manav; D. E. Brodersen; Structural conservation of the PIN domain active site across all domains of life. Protein Science 2017, 26, 1474-1492, 10.1002/pro.3193.
- Jiwei Xu; Wei Peng; Yao Sun; Xiangxi Wang; Yihui Xu; Xuemei Li; Guangxia Gao; Zihe Rao; Structural study of MCPIP1 N-terminal conserved domain reveals a PIN-like RNase. Nucleic Acids Research 2012, 40, 6957-6965, 10.1093/nar/gks359.
- Jixiao Niu; Yuling Shi; Jingyan Xue; Ruidong Miao; Shengping Huang; Tianyi Wang; Jiong Wu; Mingui Fu; Zhao-Hui Wu; USP10 inhibits genotoxic NF-κB activation by MCPIP1-facilitated deubiquitination of NEMO. The EMBO Journal 2013, 32, 3206-3219, 10.1038/emboj.2013.247.
- Jian Liang; Yasser Saad; Tianhua Lei; Jing Wang; Dongfei Qi; Qinglin Yang; Pappachan E. Kolattukudy; Mingui Fu; MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-κB signaling. Journal of Cell Biology 2010, 191, i14-i14, 10.1083/jcb1916oia14.
- Marta Muzio; Gioacchino Natoli; Simona Saccani; Massimo Levrero; Alberto Mantovani; The Human Toll Signaling Pathway: Divergence of Nuclear Factor κB and JNK/SAPK Activation Upstream of Tumor Necrosis Factor Receptor–associated Factor 6 (TRAF6). Journal of Experimental Medicine 1998, 187, 2097-2101, 10.1084/jem.187.12.2097.
- Traci M Tanaka Hall; Multiple modes of RNA recognition by zinc finger proteins. Current Opinion in Structural Biology 2005, 15, 367-373, 10.1016/j.sbi.2005.04.004.
- Katharina U. Vogel; Stephanie Edelmann; Katharina M. Jeltsch; Arianna Bertossi; Klaus Heger; Gitta A. Heinz; Jessica Zöller; Sebastian C. Warth; Kai P. Hoefig; Claudia Lohs; et al. Roquin Paralogs 1 and 2 Redundantly Repress the Icos and Ox40 Costimulator mRNAs and Control Follicular Helper T Cell Differentiation. Immunity 2013, 38, 655-668, 10.1016/j.immuni.2012.12.004.
- Hiroshi I. Suzuki; Mayu Arase; Hironori Matsuyama; Young Lim Choi; Toshihide Ueno; Hiroyuki Mano; Koichi Sugimoto; Kohei Miyazono; MCPIP1 Ribonuclease Antagonizes Dicer and Terminates MicroRNA Biogenesis through Precursor MicroRNA Degradation. Molecular Cell 2011, 44, 424-436, 10.1016/j.molcel.2011.09.012.
- Mariko Yokogawa; Takashi Tsushima; Nobuo N. Noda; Hiroyuki Kumeta; Yoshiaki Enokizono; Kazuo Yamashita; Daron M. Standley; Osamu Takeuchi; Shizuo Akira; Fuyuhiko Inagaki; et al. Structural basis for the regulation of enzymatic activity of Regnase-1 by domain-domain interactions. Scientific Reports 2016, 6, 22324, 10.1038/srep22324.
- Marc Bergdoll; Marie-Hélène Remy; Christine Cagnon; Jean-Michel Masson; Philippe Dumas; Proline-dependent oligomerization with arm exchange. Structure 1997, 5, 391-401, 10.1016/s0969-2126(97)00196-2.
- Christine Happel; Dhivya Ramalingam; Joseph M. Ziegelbauer; Virus-Mediated Alterations in miRNA Factors and Degradation of Viral miRNAs by MCPIP1. PLOS Biology 2016, 14, e2000998, 10.1371/journal.pbio.2000998.
- Shufeng Liu; Chao Qiu; Ruidong Miao; Jianhua Zhou; Aram Lee; Baoming Liu; Sandra N. Lester; Weihui Fu; Lingyan Zhu; Linxia Zhang; et al. MCPIP1 restricts HIV infection and is rapidly degraded in activated CD4+ T cells. Proceedings of the National Academy of Sciences 2013, 110, 19083-19088, 10.1073/pnas.1316208110.
- Maya Saleh; Charles O. Elson; Experimental Inflammatory Bowel Disease: Insights into the Host-Microbiota Dialog. Immunity 2011, 34, 293-302, 10.1016/j.immuni.2011.03.008.
- Ren-Jye Lin; Jan-Show Chu; Hsu-Ling Chien; Chung-Hsin Tseng; Pin-Chen Ko; Yung-Yu Mei; Wei-Chun Tang; Yu-Ting Kao; Hui-Ying Cheng; Yu-Chih Liang; et al. MCPIP1 Suppresses Hepatitis C Virus Replication and Negatively Regulates Virus-Induced Proinflammatory Cytokine Responses. The Journal of Immunology 2014, 193, 4159-4168, 10.4049/jimmunol.1400337.
- Jian Liang; Jing Wang; Yasser Saad; Logan Warble; Edilu Becerra; Pappachan E Kolattukudy; Participation of MCP-induced protein 1 in lipopolysaccharide preconditioning-induced ischemic stroke tolerance by regulating the expression of proinflammatory cytokines. Journal of Neuroinflammation 2011, 8, 182-182, 10.1186/1742-2094-8-182.
- Yun-Ji Lim; Ji-Ae Choi; Jeong-Hwan Lee; Chul Hee Choi; Hwa-Jung Kim; Chang-Hwa Song; Mycobacterium tuberculosis 38-kDa antigen induces endoplasmic reticulum stress-mediated apoptosis via toll-like receptor 2/4. Apoptosis 2014, 20, 358-370, 10.1007/s10495-014-1080-2.
- Takuya Uehata; Hidenori Iwasaki; Alexis Vandenbon; Kazufumi Matsushita; Eduardo Hernandez-Cuellar; Kanako Kuniyoshi; Takashi Satoh; Takashi Mino; Yutaka Suzuki; Daron M. Standley; et al. Malt1-Induced Cleavage of Regnase-1 in CD4+ Helper T Cells Regulates Immune Activation. Cell 2013, 153, 1036-1049, 10.1016/j.cell.2013.04.034.
- Katharina M Jeltsch; Desheng Hu; Sven Brenner; Jessica Zöller; Gitta A Heinz; Daniel Nagel; Katharina U Vogel; Nina Rehage; Sebastian C Warth; Stephanie L Edelmann; et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote TH17 differentiation. Nature Immunology 2014, 15, 1079-1089, 10.1038/ni.3008.
- Kazufumi Matsushita; Hiroki Tanaka; Koubun Yasuda; Takumi Adachi; Ayumi Fukuoka; Shoko Akasaki; Atsuhide Koida; Etsushi Kuroda; Shizuo Akira; Tomohiro Yoshimoto; et al. Regnase-1 degradation is crucial for IL-33– and IL-25–mediated ILC2 activation. JCI Insight 2020, 5, 5, 10.1172/jci.insight.131480.
- Ren-Jye Lin; Hsu-Ling Chien; Shyr-Yi Lin; Bi-Lan Chang; Han-Pang Yu; Wei-Chun Tang; Yi-Ling Lin; MCPIP1 ribonuclease exhibits broad-spectrum antiviral effects through viral RNA binding and degradation. Nucleic Acids Research 2013, 41, 3314-3326, 10.1093/nar/gkt019.
- Yisong Qian; Xiuzhen Li; Ruidong Miao; Shufeng Liu; Hong-Bo Xin; Xiaotian Huang; Tony T. Wang; Mingui Fu; Selective degradation of plasmid-derived mRNAs by MCPIP1 RNase. Biochemical Journal 2019, 476, 2927-2938, 10.1042/bcj20190646.
- Yong Li; Xuan Huang; Shengping Huang; Hui He; Tianhua Lei; Fatma Saaoud; Xiao-Qiang Yu; Ari Melnick; Anil Kumar; Christopher J Papasian; et al. Central role of myeloid MCPIP1 in protecting against LPS-induced inflammation and lung injury. Signal Transduction and Targeted Therapy 2017, 2, 17066, 10.1038/sigtrans.2017.66.
- Hongmei Li; Tony T. Wang; MCPIP1/regnase-I inhibits simian immunodeficiency virus and is not counteracted by Vpx. Journal of General Virology 2016, 97, 1693-1698, 10.1099/jgv.0.000482.
- Hidenori Iwasaki; Osamu Takeuchi; Shunsuke Teraguchi; Kazufumi Matsushita; Takuya Uehata; Kanako Kuniyoshi; Takashi Satoh; Tatsuya Saitoh; Mutsuyoshi Matsushita; Daron M Standley; et al. The IκB kinase complex regulates the stability of cytokine-encoding mRNA induced by TLR–IL-1R by controlling degradation of regnase-1. Nature Immunology 2011, 12, 1167-1175, 10.1038/ni.2137.
- Takuji Iwamoto; Hiroshi Okamoto; Yoshiaki Toyama; Shigeki Momohara; Molecular aspects of rheumatoid arthritis: chemokines in the joints of patients. The FEBS Journal 2008, 275, 4448-4455, 10.1111/j.1742-4658.2008.06580.x.
- Minoru Hasegawa; The roles of chemokines in leukocyte recruitment and fibrosis in systemic sclerosis. Frontiers in Bioscience 2008, 13, 3637-3647, 10.2741/2955.
- Vidyasaral Murugan; Michael J. Peck; SIGNAL TRANSDUCTION PATHWAYS LINKING THE ACTIVATION OF ALVEOLAR MACROPHAGES WITH THE RECRUITMENT OF NEUTROPHILS TO LUNGS IN CHRONIC OBSTRUCTIVE PULMONARY DISEASE. Experimental Lung Research 2009, 35, 439-485, 10.1080/01902140902759290.
- G. H. Tesch; MCP-1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. American Journal of Physiology-Renal Physiology 2008, 294, F697-F701, 10.1152/ajprenal.00016.2008.
- Susan E. Wozniak; Laura L. Gee; Mitchell S. Wachtel; Eldo E. Frezza; Adipose Tissue: The New Endocrine Organ? A Review Article. Digestive Diseases and Sciences 2008, 54, 1847-1856, 10.1007/s10620-008-0585-3.
- Gali Soria; Adit Ben-Baruch; The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Letters 2008, 267, 271-285, 10.1016/j.canlet.2008.03.018.
- Kangkai Wang; Jianli Niu; Hyunbae Kim; Pappachan E. Kolattukudy; Osteoclast precursor differentiation by MCPIP via oxidative stress, endoplasmic reticulum stress, and autophagy. Journal of Molecular Cell Biology 2011, 3, 360-368, 10.1093/jmcb/mjr021.
- Jingjing Da; Ming Zhuo; Minzhang Qian; MCPIP is induced by cholesterol and participated in cholesterol-caused DNA damage in HUVEC. International journal of clinical and experimental pathology 2015, 8, 10625-10634, .
- Dongfei Qi; Shengping Huang; Ruidong Miao; Zhi-Gang She; Tim Quinn; Yingzi Chang; Jianguo Liu; Daping Fan; Y. Eugene Chen; Mingui Fu; et al. Monocyte Chemotactic Protein-induced Protein 1 (MCPIP1) Suppresses Stress Granule Formation and Determines Apoptosis under Stress. Journal of Biological Chemistry 2011, 286, 41692-41700, 10.1074/jbc.m111.276006.
- Emmanuel George Vrotsos; Pappachan E. Kolattukudy; Kiminobu Sugaya; MCP-1 involvement in glial differentiation of neuroprogenitor cells through APP signaling. Brain Research Bulletin 2009, 79, 97-103, 10.1016/j.brainresbull.2009.01.004.
- Jianli Niu; Asim Azfer; Olga Zhelyabovska; Sumbul Fatma; Pappachan E. Kolattukudy; Monocyte Chemotactic Protein (MCP)-1 Promotes Angiogenesis via a Novel Transcription Factor, MCP-1-induced Protein (MCPIP). Journal of Biological Chemistry 2008, 283, 14542-14551, 10.1074/jbc.m802139200.
- Craig W. Younce; Asim Azfer; Pappachan E. Kolattukudy; MCP-1 (Monocyte Chemotactic Protein-1)-induced Protein, a Recently Identified Zinc Finger Protein, Induces Adipogenesis in 3T3-L1 Pre-adipocytes without Peroxisome Proliferator-activated Receptor γ. Journal of Biological Chemistry 2009, 284, 27620-27628, 10.1074/jbc.m109.025320.
- Ana G. Cristancho; Mitchell A. Lazar; Forming functional fat: a growing understanding of adipocyte differentiation. Nature Reviews Molecular Cell Biology 2011, 12, 722-734, 10.1038/nrm3198.
- Barbara Lipert; Paulina Węgrzyn; Henrike Sell; Juergen Eckel; Marek Winiarski; Andrzej Budzyński; Maciej Matlok; Jerzy Kotlinowski; Lindsay Ramage; Maciej Malecki; et al. Monocyte chemoattractant protein-induced protein 1 impairs adipogenesis in 3T3-L1 cells. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2014, 1843, 780-788, 10.1016/j.bbamcr.2014.01.001.
- Magdalena Losko; Agata Lichawska-Cieslar; Maria Kulecka; Agnieszka Paziewska; Izabela Rumienczyk; Michal Mikula; Jolanta Jura; Ectopic overexpression of MCPIP1 impairs adipogenesis by modulating microRNAs. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2018, 1865, 186-195, 10.1016/j.bbamcr.2017.09.010.
- Kyung Hee Hong; Jewon Ryu; Ki Hoon Han; Monocyte chemoattractant protein-1–induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood 2005, 105, 1405-1407, 10.1182/blood-2004-08-3178.
- Anna Labedz-Maslowska; Barbara Lipert; Dominika Berdecka; Sylwia Kedracka-Krok; Urszula Jankowska; Elzbieta Kamycka; Malgorzata Sekula; Zbigniew Madeja; Buddhadeb Dawn; Jolanta Jura; et al. Monocyte Chemoattractant Protein-Induced Protein 1 (MCPIP1) Enhances Angiogenic and Cardiomyogenic Potential of Murine Bone Marrow-Derived Mesenchymal Stem Cells. PLoS ONE 2015, 10, e0133746, 10.1371/journal.pone.0133746.
- Janusz Ligeza; Paulina Marona; Natalia Gach; Barbara Lipert; Katarzyna Miekus; Waclaw Wilk; Janusz Jaszczynski; Andrzej Stelmach; Agnieszka Loboda; Jozef Dulak; et al. MCPIP1 contributes to clear cell renal cell carcinomas development. Angiogenesis 2017, 20, 325-340, 10.1007/s10456-017-9540-2.