Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Amongst the consequences of spinal cord injury (SCI) on the central nervous system, the loss of inhibition is a common finding, albeit not always observed, and it is likely to fluctuate over time. Changes in cortical excitability involve a plethora of mechanisms, which individual effects may combine in complex and variable outcomes on the process of functional and structural recovery.
- cortical inhibition
- spinal cord injury
- neocortex
- disinhibition
- interneuron
1. Introduction: SCI Harms the Brain
Traumatic spinal cord injury (SCI) is a sudden and unpredictable incident that destroys portions of the spinal cord, leading to motor and sensory deficits, as well as dysfunctions of the somatic and autonomic nervous systems [1]. Beyond the loss of movement control, typical deficits include the loss of bladder and bowel control, declined sexual functions and chronic pain, among others [1,2]. SCI can occur at any age, and the damage is irreversible. However, constant improvements in healthcare and treatment, as well as increased awareness about the needs of patients over the last century, have significantly ameliorated the quality of life and lifespan following SCI [3,4,5,6,7,8,9]. Thus, it is even more pressing to identify interventions enabling the recovery of functions lost after SCI. The recovery of muscle control is a crucial element to improve the quality of life and the autonomy of SCI patients. Accordingly, rehabilitation and active lifestyle have been recognized as crucial processes that help to regain independence and to reduce health complications resulting from prolonged inactivity [10,11,12,13,14]. Nevertheless, the timely implementation of efficient strategies remains often neglected, affecting motor recovery and, together with accompanying morbidities, decreasing the likelihood of returning to a fully independent life routine [15,16]. Furthermore, various therapies addressing the symptoms of SCI are being developed with promising outcomes for management and reduction in secondary damage, increased neuroprotection and improved neuroregeneration [5,17]. However, despite constant improvements, an effective cure, leading to major functional recovery based on the regeneration of neuronal connectivity across the lesion, is still missing [17,18,19,20].
Many of the neurons that become disconnected following SCI reside outside the spinal cord, such as the motor neurons of the primary motor cortex, which are crucial for the control of voluntary movements. These disconnected neurons are a resource for the long-term regeneration and functional recovery of the central nervous system. However, the axotomy resulting from SCI has an impact on the physiology of the cortical and corticospinal network [21,22,23], which can complicate or even hinder the recovery process. Several attempts to address the clinical symptoms of SCI have therefore explored the possibility to retune neuronal activity in the corticospinal network [24,25]. Finding ways to reconnect cortical motor neurons to their original targets constitutes a daunting task. In addition, early assessments of the severity of SCI, especially in an acute situation, are difficult and inherently inaccurate [26]. This lack of knowledge is a major hurdle for the design of an effective and patient-specific treatment. Therefore, the assessment of cortical activity in SCI patients, for example, using electro-encephalography (EEG), transcranial magnetic stimulation (TMS), etc., has been extremely convenient, as it relies on non-invasive techniques [27,28]. In addition, animal models are available to resolve the molecular mechanisms of brain dysfunction after SCI.
2. Possible Causes for the Loss of Inhibition
Pre-clinical research helps to define mechanisms that converge to an altered ratio of excitation and inhibition following SCI and to refine hypotheses about the reasons for phenotypic heterogeneity, as reported in clinical findings. Focusing on molecular “bottlenecks” of inhibitory neurotransmission, the likely mechanisms of cortical disinhibition after SCI are presented in the following section (Figure 1).
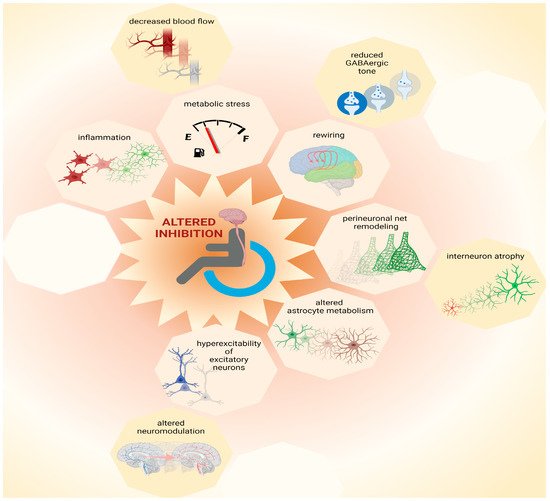
Figure 1. Possible causes for loss of inhibition. Multiple mechanisms contribute to network plasticity and altered inhibition in cortical and subcortical areas after SCI. These include metabolic stress, which can be exacerbated by inflammation and by decreased blood flow, as well as altered astrocytic metabolism. Furthermore, increased excitability of principal (excitatory) neurons may be exacerbated by altered neuromodulation, contributing to increased output volume in corticospinal circuits. Moreover, disinhibition is associated with decreased GABAergic tone that contributes to plasticity during network rewiring. Additionally, remodeling of perineuronal nets, which involves atrophy of interneurons, can contribute to complex patterns of altered inhibition in the central nervous system after SCI. Thus, multiple mechanisms, some of which are yet to be identified (represented by empty octagons) may coexist and combine heterogeneously amongst each other and/or to other pathophysiological components, increasing inter-patient variability. This figure was created with biorender.com.
2.1. Metabolic Stress
The synthesis of neurotransmitters, such as glutamate and GABA, demand considerable energy consumption [93]. Of these, GABAergic neurotransmission has the highest demand. For this reason, inhibitory neurons such as parvalbumin-positive and axo-axonic interneurons are vulnerable and respond to stress with decreased GABAergic neurotransmission [94]. After SCI, the axotomy of corticospinal neurons may cause stress, spreading from the spinal cord to cortical areas [21,22,23]. Additional sources of stress may be inflammation [95] and impaired cerebral blood flow [96]. Furthermore, stress may coincide with high metabolic demand due to increased cortical excitability [22]. Hence, multiple stressors may affect interneurons, as also suggested by the direct observation of their atrophy after SCI [32]. The alteration in brain network performance as a consequence of dysfunctional interneurons may impact cognition negatively [97,98], such as occasionally documented in SCI patients [99].
2.2. Inflammation
Microglia activation plays a pivotal role in SCI. Furthermore, chronic inflammation seems to affect supraspinal regions as well [100]. This is not surprising, given the axotomy of several cortical and subcortical neurons [21,22,23] and the continuity of the spinal cord and brain. Regrettably, although the wave of inflammation after SCI has been well time-resolved near the lesion site [101,102,103], little is known about the spatial distribution of immune cell activation across the CNS following various types of injury and treatment. Thus, it is hard to define which neurons are most exposed to inflammatory processes and which are spared and exclusively undergo plasticity processes independent of inflammation. Among several disinhibitory effects [104], activated microglia affect the activity of proteins that transport chloride across the neuronal cell membrane and cause the dissipation of chloride trans-membrane gradients with consequent reductions in the GABAergic inhibitory drive [105]. Decreased GABAergic drive causes the neurotransmitter GABA to be intrinsically less effective in hyperpolarizing the neuronal membrane to mediate inhibition. The dissipation of chloride gradients upon microglia activation after SCI may therefore contribute to disinhibition as well, as documented in other pathologies [105,106].
2.3. Remodeling of Perineuronal Nets
The extracellular elements of proteoglycans, known as perineuronal nets, mediate stability in network architecture as well as protection from stress [107]. Dismantling perineuronal nets promotes plasticity and network remodeling [108], which are pertinent for recovery after SCI [109,110]. Furthermore, perineuronal nets are closely associated with specific categories of cortical interneurons [111,112]. Thereby, these extracellular elements can control neuronal synaptic connectivity and intrinsic firing properties [112,113]. Thus, the dismantling of perineuronal nets can affect the activity and connectivity of interneurons, determining altered cortical inhibition after SCI. Strikingly, the advantage of increased plasticity derived from the dismantling of perineuronal nets comes along with the challenge presented by increased metabolic stress. Indeed, perineuronal nets limit excitotoxicity by sheltering synaptic contacts and reducing oxidative stress to which interneurons are most vulnerable [94,114,115]. Since neocortical perineuronal nets undergo remodeling after SCI that is associated with the atrophy of interneurons [32], these extracellular elements may be involved in both beneficial and detrimental aspects of cortical disinhibition.
2.4. Altered Astrocyte Metabolism and Physiology
Axotomy and neuronal trauma perturb the physiology of astrocytes [116]. Trauma suffered by cortical principal neurons upon axotomy [21,22,23] may therefore alter signaling from astrocytes to surrounding neurons. Astrocytes can release neuromodulators controlling neuronal activity by gliotransmission [117,118,119]. Moreover, astrocytes are crucial for the metabolism of neurotransmitters [120,121]. Thus, under pathological conditions, altered astrocytic activity may affect neurotransmission, and in particular GABAergic inhibition, via altered metabolic support as well [122]. The involvement of astrocytes in SCI-derived pathology has been widely studied [123], albeit not in cerebral regions.
2.5. Rewiring of Cortical Circuits
A physiological process closely tied to disinhibitory mechanisms is the rewiring of cortical circuits after SCI. Like other injuries involving deafferentation of the central nervous system, SCI causes the rewiring of cortical and subcortical areas, as well as the shift of somatotopic representations and changes in competence of motor areas, which relay on significant events of network plasticity [124]. Decreases in GABAergic inhibition appear crucial for network remodeling [125,126] because plasticity and learning are tightly related to changes in cortical GABA [127]. For instance, reduced GABAergic inhibition causes qualitative and quantitative changes in the inducibility of long term potentiation in the neocortex [128,129]. Furthermore, reduced GABAergic tone, i.e., diminished concentrations of extrasynaptic GABA, supports motor plasticity and motor recovery in various types of pathology [126,130]. Thus, several mechanisms that contribute to reduced GABAergic inhibition in the brain after SCI may also partake into the process of cortical rewiring and recovery.
2.6. Hyperexcitability of Excitatory Neurons
Some causes of hyperexcitability may be independent from disinhibition. Nevertheless, solving pathological hyperexcitability may necessitate retuning inhibitory components. For instance, axotomy results in the depolarization and hyperexcitability of cortical principal neurons after SCI [22]. The retuning of hyperexcitable neurons may benefit from increased inhibition, for instance, through GABAB receptor signaling, which controls dendritic excitability via intracellular calcium signaling [131]. In contrast, disinhibition has additive exacerbating effects on the intrinsic hyperexcitability of principal neurons. Furthermore, other types of neuromodulation that exert inhibitory control on CNS neurons, e.g., the ones mediated by serotonin, are impaired after SCI [132]. The activation of serotonin receptors has been directly shown to control the excitability of cortical neurons [133]. Additionally, the enrichment of serotonin receptors at the axonal hillock of layer V pyramidal neurons implies a crucial role in the control of cortical functional output [134]. Thus, non-GABAergic neuromodulation can directly control the excitability of principal neurons as well as the excitability of interneurons [135,136]. Since altered serotoninergic neuromodulation can affect directly and indirectly the volume of corticospinal output, serotonin neuromodulation may be a key component in controlling cortical output after SCI. Strikingly, controlling serotoninergic neuromodulation has already proven to support better recovery after SCI [137,138,139].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23105622
This entry is offline, you can click here to edit this entry!