Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The tripartite motif (TRIM) gene family is a large group of E3 ubiquitin ligase proteins that can also have proteasome-independent functions. TRIM/RBCC are a large family of proteins that include more than 80 proteins, most of which act as E3 ligases and catalyze the direct transfer of Ubiquitin, SUMO and ISG15 on specific protein substrates. They are involved in oncogenesis processes and in cellular immunity.
- TRIM proteins
- TRIM8
- p53
- cancer
1. TRIM Proteins Biological Functions
TRIM proteins are involved in distinct cellular processes, despite showing a similar structure: regulation of cellular homeostasis, cell cycle, senescence, apoptosis, differentiation, specific metabolic pathways, meiosis and protein quality control [1][2][3]. They exert their actions on transcriptional regulation, cytoskeletal remodeling, intracellular trafficking, membrane repair and oncogenesis [4][5][6][7]. Moreover, these proteins are implicated in the development and regulation of the immune system [8][9][10].
Most TRIM molecules act as E3 ligases and directly catalyze the transfer of Ub, SUMO and ISG15 on specific protein substrates [11][12][13][14]. The conjugation reaction of ubiquitin to a substrate is catalyzed by E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzymes and E3 ubiquitin ligases [15]. The E3 ubiquitin ligases can be divided into two major classes: the homologous to E6-AP COOH terminus (HECT) E3 ubiquitin ligase family and the RING-finger-containing E3 ubiquitin ligase family [16][17]. The high number of E3 ligases is associated with their specificity in selectively targeting protein substrates [1]. Mostly, the enzymatic activity exerted by the E3 ligases on ubiquitin and Ubiquitin-like molecules (UBL) depends on the presence in the protein structure of the RING domain [18][19]. E3 ligases transfer the ubiquitin or UBLs from E2 conjugating enzymes to the substrates, and thus they are responsible for recognizing the substrates and are determinants of target specificity [20]. In order for ubiquitination to take place, however, it is not sufficient for the RING domain to recognize the specific substrate, but specific functional protein dimers must be formed [21][22][23]. The B-box and coiled-coil domains are responsible for the dimerization of TRIM proteins [24][25].
Many TRIM proteins play a pivotal role during mitosis and cell-cycle progression. Specifically, TRIM19, TRIM22, TRIM28, TRIM37 and TRIM6 are important during prophase; TRIM19, TRIM32 and TRIM69 in prometaphase; TRIM17, TRIM36 and TRIM69 in metaphase; and TRIM17, TRIM21, TRIM47 and TRIM76 in cytokinesis. Furthermore, in the course of the bipolar spindle assembly during all phases of mitosis, TRIM8 is involved in the mitotic spindle formation through interaction with two important regulators of mitotic spindle machinery and cytoskeleton reorganization, KIF11 and KIFC1, and through localization at the mitotic spindle [26]. TRIM8 colocalizes on centrosomes with Plk1 and straight reacts to CEP170-like protein. This interaction, suppressing TRIM8 function, induces a delay of the mitosis progression with a cell accumulation in the G2/M phase. TRIM8 is also necessary for chromosomal stability. As a matter of fact, the suppressing of TRIM8 induced an increased rate of chromosomal instability leading to a significant rise of cells with less than 46 chromosomes [1][16].
TRIM proteins have the distinctive feature of exerting a large variety of different roles and activities because of their ubiquitination or ubiquitin-like function that labels the target proteins to be degraded at the proteasome level, as well as stabilize or dislocate them in various cellular compartments through such modifications. Ubiquitination is a post-transductional modification of protein substrates necessary for different biological mechanisms, such as:
-
Degradation of toxic protein aggregates [29];
The alteration of the post-transduction mechanism of ubiquitination affects the functionality of protein substrates, with consequent alteration of the biological mechanisms in which they are involved. At a macroscopic level, these alterations can lead to the development of various pathological conditions, including tumor pathologies [24][31][32]. TRIM proteins are involved in carcinogenesis. In particular, these proteins are implicated in several biological functions: DNA repair, metastasis, tumor-suppressive and oncogenic regulation [4]. Furthermore, some of the TRIM family proteins play a pivotal role in autophagy and innate immunity and regulate important cellular processes, such as intracellular signaling and transcription [16]. The down-regulation or overexpression of TRIM proteins has long been investigated in the study of oncogenesis. However, many reports showed that TRIM alterations were observed in lung cancer, breast cancer, liver cancer, colorectal cancer and prostate cancer [33][34]. Indeed, reduced expression of these proteins could reflect the suppressive role of the tumor, whereas their over-expression could reflect their contribution to the disease development and/or progression. Therefore, some TRIMs could be considered biomarkers for some kind of cancer. In particular, TRIM11, TRIM14, TRIM24, TRIM25, TRIM27, TIM28, TRIM29, TRIM33, TRIM37, TRIM44 and TRIM59 are the most associated with cancer [33].
2. TRIM Proteins and Cancer Pathogenesis
TRIM proteins could influence cancer pathogenesis through the following mechanisms:
-
Chromosomal translocation [35]. It could generate a fusion protein without activity or with a different activity that could dysregulate some signaling pathways, leading to the generation of some tumor shapes. An example is a translocation between the TRIM19 gene (PML) on chromosome 15 and the retinoic acid receptor α (RARa) gene on chromosome 17. This translocation leads to the formation of a fusion protein that represses acid signaling retinoic and is associated with Acute Promyelocytic Leukemia [36]. Such similar examples are the following: TRIM24, TRIM27 and TRIM33 were found in translocations with the RET gene and are involved in papillary thyroid cancer, lymphoma and non-small cell lung carcinoma, respectively. Similarly, TRIM24 was found translocated with the BRAF gene in melanoma and lung cancer and with the FGFR1 gene in myeloproliferative syndrome [33];
-
Modulation of the activity and stability of p53. TRIM11, TRIM13, TRIM21, TRIM24, TRIM25, TRIM28, TRIM29, TRIM31, TRIM32, TRIM39 and TRIM59 can ubiquitinate the p53 protein, a fundamental macromolecule in cell development whose purpose is to promote genomic stability and induce cell cycle arrest and apoptosis if extensive DNA damage is found in the cell. The ubiquitination of this protein leads to its direct degradation or to its sequestration in the cytoplasm: since it can no longer penetrate the nucleus, the ubiquitinated protein is no longer able to detect any damage to the DNA; consequently, the cell replicates itself by transmitting the same error in the nucleic acid sequence, resulting in the possible onset of tumor forms [37];
-
Regulation of pathways to cancer stemness, including STAT signaling, AKT signaling, NANOGSox2-Oct-3/4 networks. Specifically, through these pathways, TRIM28 is involved in breast cancer, TRIM24 in glioblastoma and colorectal cancer, TRIMs 14 in gastric cancer and TRIM16 has been associated as a negative regulator of stemness in breast and ovarian cancer cells [33].
Stem cells (SCs) are cells with no signs of differentiation, capable of self-renewing and generating progeny capable of differentiating into different cell types. They constitute the reserve elements of human tissues; in fact, they are activated only to restore tissue damage or to ensure normal cell turnover. SCs are capable of self-renewal, are multi-potent and immortal, and are highly resistant to chemical and physical agents, all characteristics also possessed by cancer cells. Furthermore, SCs tend to maintain the ability to de-differentiate in order to return to a primitive state of development. Such cells cannot survive outside their environment or in case of deficiency of specific cytokines and growth factors. Mutated stem cells, however, despite having all the aspects of stem cells, are unable to support tissue homeostasis, favoring, instead, the onset and progression of tumor diseases. The stem characteristics common to HF and cancer cells provide the building blocks for cancer maintenance and survival, from the potential for self-renewal and differentiation to the organization of microenvironments that support stemness. Thus, cancer stem cells (CSCs) are defined as the small population of cells within tumors that possess stem properties that support cancer development, such as advanced capabilities for cloning, growth, metastasis, re-proliferation and self-renewal. CSCs exhibit remarkable organizational skills. In fact, they can educate neighboring cells to provide nutrients and collaborate in evading the immune system, thus creating an environment favorable for tumor progression [38]. CSCs give rise to heterogeneous cell populations, often with a high potential for plasticity, high resistance to stressors, such as low oxygen or nutrient levels, or the initiation of cell death by chemotherapeutic agents, and the capability for quiescence as a typical response to these factors [39]. Among the possible pathways by which TRIM proteins act on tumor stemness are the signaling of STAT, AKT (Figure 1) and the NANOGSox2-Oct-3/4 networks [40]. In fact, TRIM proteins control stem cell characteristics mostly positively by enhancing the activity of core transcription factors, induction of specific signaling pathways, epigenetic silencing of pro-differentiation genes, metabolic reprogramming and activation of the epithelium-mesenchymal transition pathway. Several members, such as TRIM24 and TRIM28, negatively regulate stem cell self-renewal, presumably by ubiquitin-mediated degradation of stem cell transcription factors or inhibition of specific signaling pathways. Furthermore, several TRIM proteins, such as TRIM8, modulate the stem cell phenotype both positively and negatively [38]. In addition, the role of TRIM proteins on autophagic processes may represent a not-well investigated mechanism involved in cancer stemness [33].
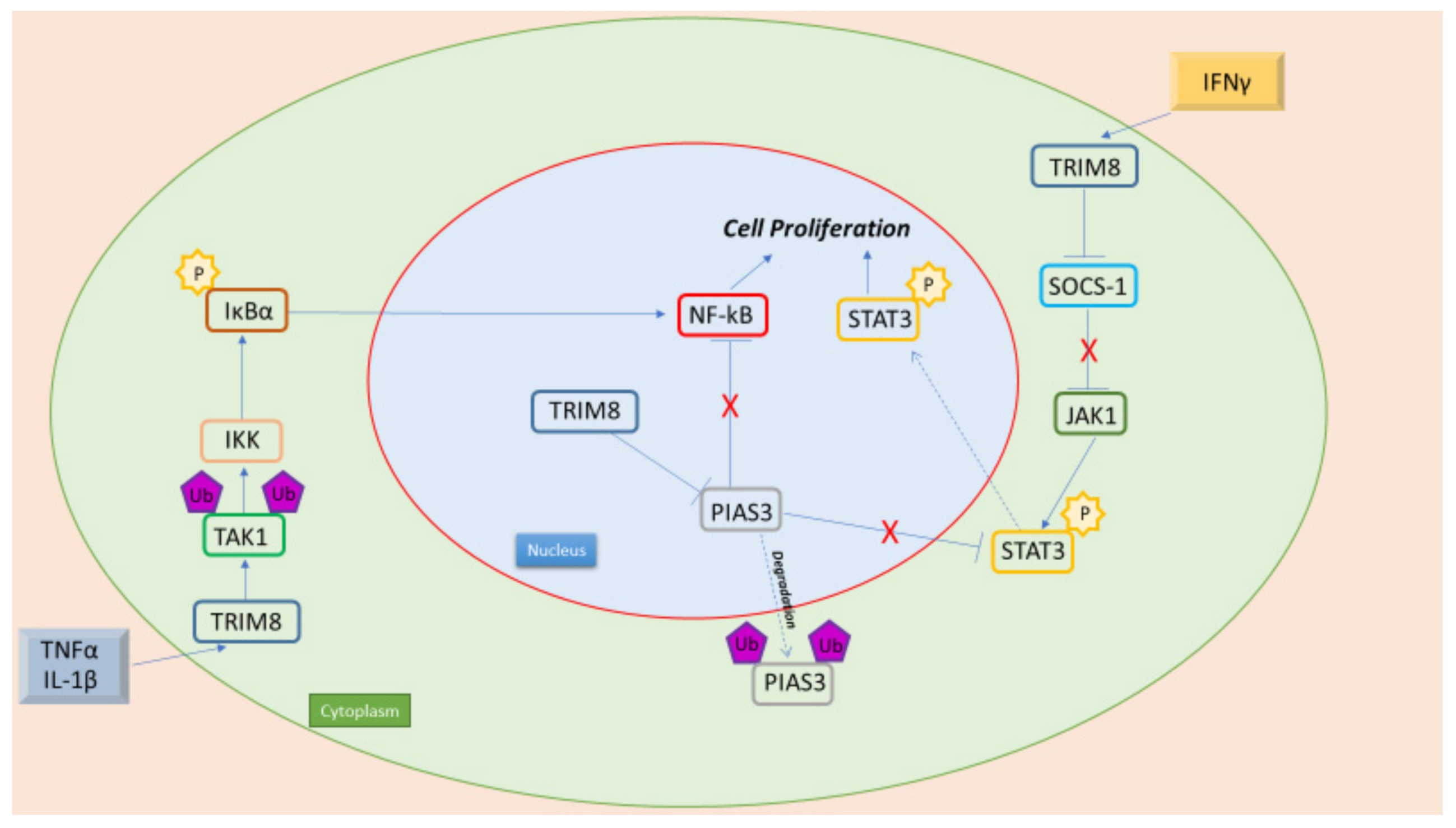
Figure 1. TRIM8 is an oncogenic protein, with its interaction with NF-kB and STAT3 leading to cell proliferation. Pro-inflammatory cytokines (TNFα e IL-1β) promote NF-kB activation through TRIM8. In fact, TRIM8 promotes TAK1 Lys63- linked polyubiquitination, leading to IKK kinase activation. Moreover, in the nucleus, TRIM8 promotes the translocation of PIAS3 in the cytoplasm, where it is then degraded. PIAS3 in the nucleus interacts with NF-kB preventing its activation. Furthermore, TRIM8 induces the activation of the JAK-STAT pathway promoted by IFN-γ through the degradation of two STAT protein inhibitors, PIAS3 and SOCS-1.
The epithelium-mesenchymal transition (EMT) is a biological process that allows a polarized epithelial cell, which normally reacts with the basement membrane through its basal surface, to suffer several biochemical changes that permit it to acquire a mesenchymal cell phenotype, which includes a prominent migratory ability, invasiveness, significant opposition to apoptosis and considerably raised production of components of the extracellular matrix. The achievement of an EMT is signaled by the degradation of the underlying basement membrane and the development of a mesenchymal cell that can move away from the epithelial layer where it arises [42].
The biological processes that initiate an EMT include activation of specific transcription factors, expression of cell surface proteins, reorganization and expression of cytoskeletal proteins, production of degradative enzymes and changes in the expression of specific microRNAs. Some of these factors can be used as biomarkers to establish the switch of a cell through an EMT [43].
This entry is adapted from the peer-reviewed paper 10.3390/cancers14092309
References
- Marzano, F.; Guerrini, L.; Pesole, G.; Sbisà, E.; Tullo, A. Emerging Roles of TRIM8 in Health and Disease. Cells 2021, 10, 561.
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villán, E.; García-Sastre, A.; Gack, M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012, 8, e1003059.
- Uchil, P.D.; Quinlan, B.D.; Chan, W.T.; Luna, J.M.; Mothes, W. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 2008, 4, e16.
- Uchil, P.D.; Hinz, A.; Siegel, S.; Coenen-Stass, A.; Pertel, T.; Luban, J.; Mothes, W. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 2013, 87, 257–272.
- Venuto, S.; Monteonofrio, L.; Cozzolino, F.; Monti, M.; Appolloni, I.; Mazza, T.; Canetti, D.; Giambra, V.; Panelli, P.; Fusco, C.; et al. TRIM8 interacts with KIF11 and KIFC1 and controls bipolar spindle formation and chromosomal stability. Cancer Lett. 2020, 473, 98–106.
- Toniato, E.; Chen, X.P.; Losman, J.; Flati, V.; Donahue, L.; Rothman, P. TRIM8/GERP RING finger protein interacts with SOCS-1. J. Biol. Chem. 2002, 277, 37315–37322.
- Dang, X.; He, B.; Ning, Q.; Liu, Y.; Chang, Y.; Chen, M. Suppression of TRIM8 by microRNA-182-5p restricts tumor necrosis factor-α-induced proliferation and migration of airway smooth muscle cells through inactivation of NF-Κb. Int. Immunopharmacol. 2020, 83, 106475.
- McNab, F.W.; Rajsbaum, R.; Stoye, J.P.; O’Garra, A. Tripartite-motif proteins and innate immune regulation. Curr. Opin. Immunol. 2011, 23, 46–56.
- Sato, T.; Okumura, F.; Kano, S.; Kondo, T.; Ariga, T.; Hatakeyama, S. TRIM32 promotes neural differentiation through retinoic acid receptor-mediated transcription. J. Cell Sci. 2011, 124 Pt 20, 3492–3502.
- Schwamborn, J.C.; Berezikov, E.; Knoblich, J.A. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 2009, 136, 913–925.
- D’Amico, F.; Mukhopadhyay, R.; Ovaa, H.; Mulder, M. Targeting TRIM Proteins: A Quest towards Drugging an Emerging Protein Class. Chembiochem 2021, 22, 2011–2031.
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920.
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8, e42426.
- Zou, W.; Zhang, D.E. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 2006, 281, 3989–3994.
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311.
- Bhaduri, U.; Merla, G. Rise of TRIM8: A Molecule of Duality. Mol. Ther. Nucleic Acids 2020, 22, 434–444.
- Iwai, K. Discovery of linear ubiquitination, a crucial regulator for immune signaling and cell death. FEBS J. 2021, 288, 1060–1069.
- Brazee, P.; Dada, L.A.; Sznajder, J.I. Role of Linear Ubiquitination in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 761–768.
- Elton, L.; Carpentier, I.; Verhelst, K.; Staal, J.; Beyaert, R. The multifaceted role of the E3 ubiquitin ligase HOIL-1: Beyond linear ubiquitination. Immunol. Rev. 2015, 266, 208–221.
- Bell, J.L.; Malyukova, A.; Holien, J.K.; Koach, J.; Parker, M.W.; Kavallaris, M.; Marshall, G.M.; Cheung, B.B. TRIM16 acts as an E3 ubiquitin ligase and can heterodimerize with other TRIM family members. PLoS ONE 2012, 7, e37470.
- Hofmann, T.G.; Möller, A.; Sirma, H.; Zentgraf, H.; Taya, Y.; Dröge, W.; Will, H.; Schmitz, M.L. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 2002, 4, 1–10.
- Ikeda, F.; Dikic, I. Atypical ubiquitin chains: New molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008, 9, 536–542.
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010, 141, 315–330.
- Gushchina, L.V.; Kwiatkowski, T.A.; Bhattacharya, S.; Weisleder, N.L. Conserved structural and functional aspects of the tripartite motif gene family point towards therapeutic applications in multiple diseases. Pharmacol. Ther. 2018, 185, 12–25.
- Yang, L.; Xia, H. TRIM Proteins in Inflammation: From Expression to Emerging Regulatory Mechanisms. Inflammation 2021, 44, 811–820.
- Venuto, S.; Castellana, S.; Monti, M.; Appolloni, I.; Fusilli, C.; Fusco, C.; Pucci, P.; Malatesta, P.; Mazza, T.; Merla, G.; et al. TRIM8-driven transcriptomic profile of neural stem cells identified glioma-related nodal genes and pathways. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 491–501.
- Venuto, S.; Merla, G. E3 Ubiquitin Ligase TRIM Proteins, Cell Cycle and Mitosis. Cells 2019, 8, 510.
- Roy, M.; Tomar, D.; Singh, K.; Lakshmi, S.; Prajapati, P.; Bhatelia, K.; Gohel, D.; Singh, R. TRIM8 regulated autophagy modulates the level of cleaved Caspase-3 subunit to inhibit genotoxic stress induced cell death. Cell. Signal. 2018, 48, 1–12.
- Maarifi, G.; Smith, N.; Maillet, S.; Moncorgé, O.; Chamontin, C.; Edouard, J.; Sohm, F.; Blanchet, F.P.; Herbeuval, J.P.; Lutfalla, G.; et al. TRIM8 is required for virus-induced IFN response in human plasmacytoid dendritic cells. Sci. Adv. 2019, 5, eaax3511.
- Bai, X.; Zhang, Y.L.; Liu, L.N. Inhibition of TRIM8 restrains ischaemia-reperfusion-mediated cerebral injury by regulation of NF-κB activation associated inflammation and apoptosis. Exp. Cell Res. 2020, 388, 111818.
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626.
- Watanabe, M.; Hatakeyama, S. TRIM proteins and diseases. J. Biochem. 2017, 161, 135–144.
- Mandell, M.A.; Saha, B.; Thompson, T.A. The Tripartite Nexus: Autophagy, Cancer, and Tripartite Motif-Containing Protein Family Members. Front. Pharmacol. 2020, 11, 308.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Shim, H.S.; Kenudson, M.; Zheng, Z.; Liebers, M.; Cha, Y.J.; Hoang Ho, Q.; Onozato, M.; Phi Le, L.; Heist, R.S.; Iafrate, A.J. Unique Genetic and Survival Characteristics of Invasive Mucinous Adenocarcinoma of the Lung. J. Thorac. Oncol. 2015, 10, 1156–1162.
- Cambiaghi, V.; Giuliani, V.; Lombardi, S.; Marinelli, C.; Toffalorio, F.; Pelicci, P.G. TRIM proteins in cancer. Adv. Exp. Med. Biol. 2012, 770, 77–91.
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120.
- Jaworska, A.M.; Wlodarczyk, N.A.; Mackiewicz, A.; Czerwinska, P. The role of TRIM family proteins in the regulation of cancer stem cell self-renewal. Stem. Cells 2020, 38, 165–173.
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem. Cells Int. 2017, 2017, 5619472.
- Czerwińska, P.; Shah, P.K.; Tomczak, K.; Klimczak, M.; Mazurek, S.; Sozańska, B.; Biecek, P.; Korski, K.; Filas, V.; Mackiewicz, A.; et al. TRIM28 multi-domain protein regulates cancer stem cell population in breast tumor development. Oncotarget 2017, 8, 863–882.
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483.
- Tse, J.C.; Kalluri, R. Mechanisms of metastasis: Epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J. Cell. Biochem. 2007, 101, 816–829.
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428.
This entry is offline, you can click here to edit this entry!