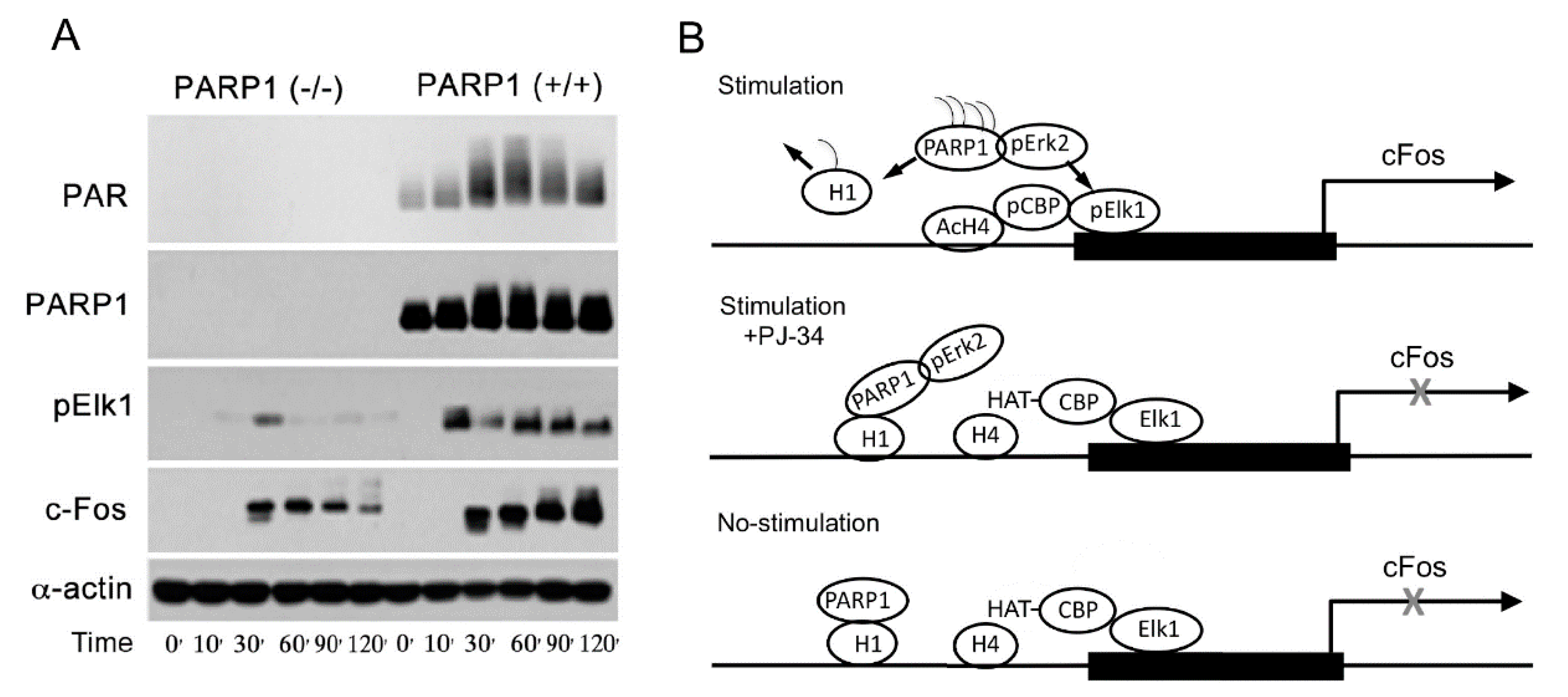
PolyADP-ribosylation is an evolutionary conserved, reversible post-translational modification of proteins. Numerous nuclear proteins act as substrates of the abundant nuclear polyADP-ribose polymerase 1 (PARP1). In this modification, negatively charged ADP-ribose chains constructed on chromatin-bound proteins, cause their repulsion from the negatively charged DNA. In accordance, polyADP-ribosylation is a post-translational modification of proteins that causes relaxation of the highly condensed structure of the chromatin. Histone H1, which is bound to the linker DNA, located between the nucleosomes, is a prominent substrate of PARP1.
- PARP1 activation
- H1 polyADP-ribosylation
- signal transduction pathways
1. Introduction
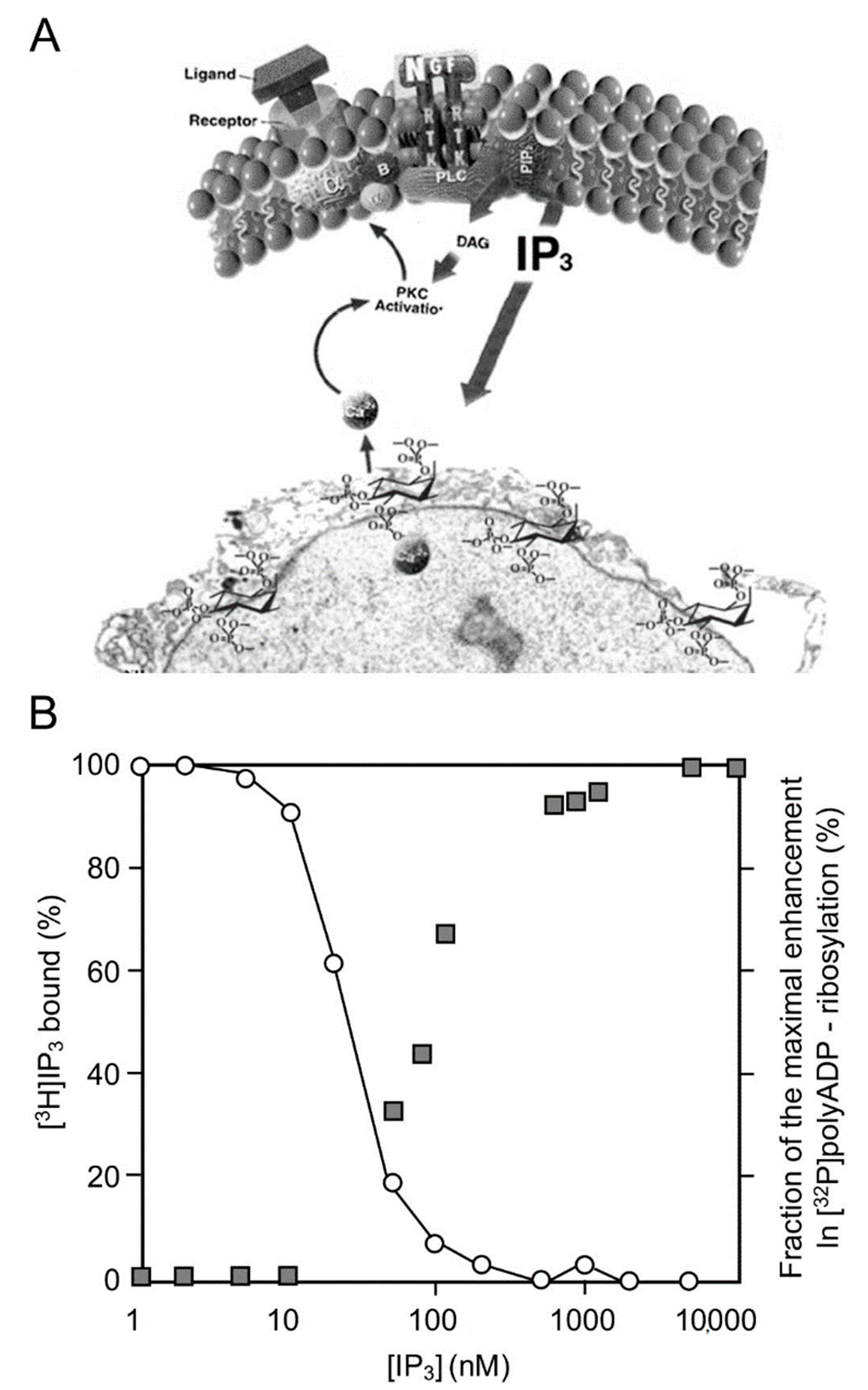
2. PARP1 Activation by Ca2+ Released from Intracellular IP3-Gated Stores
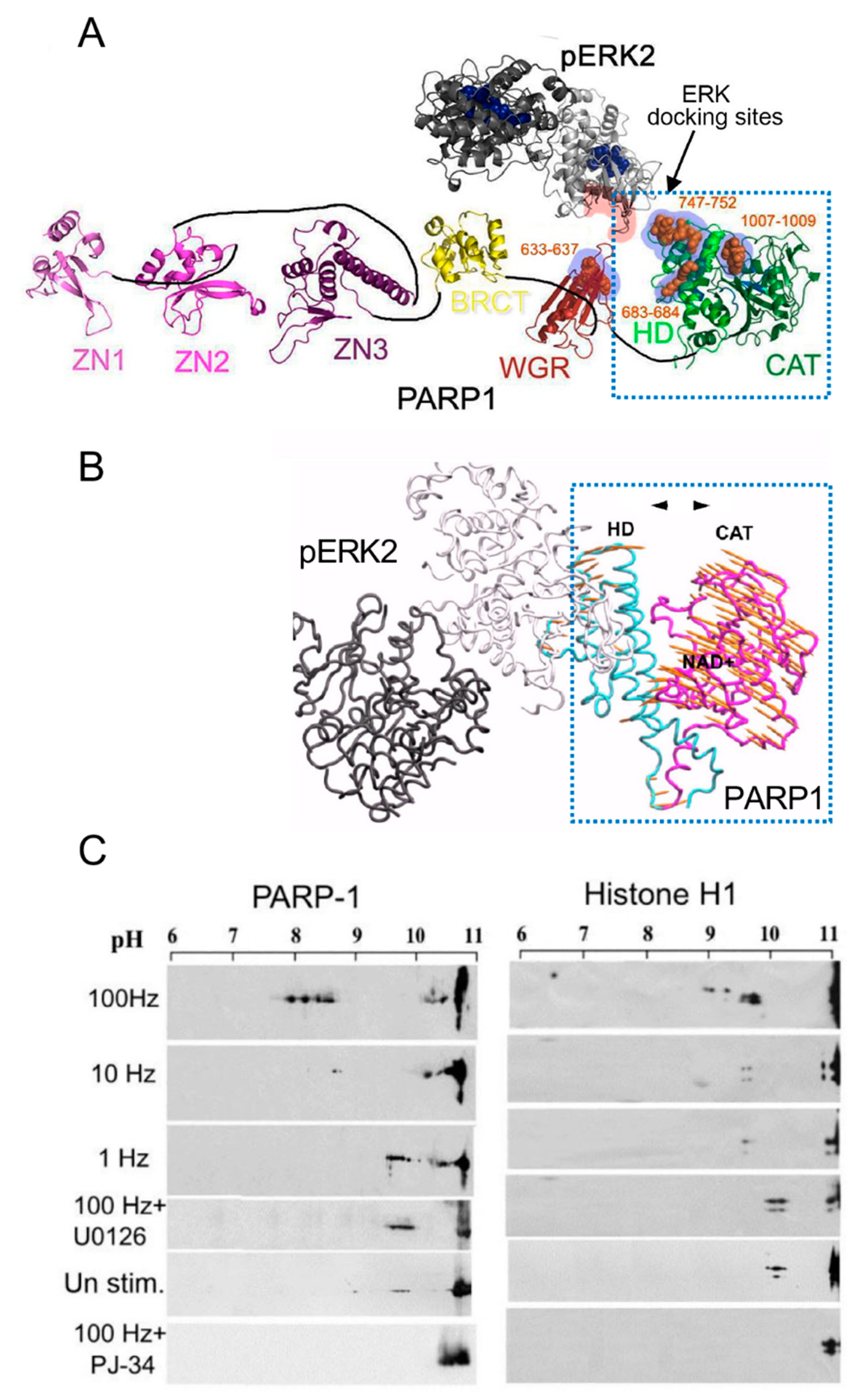
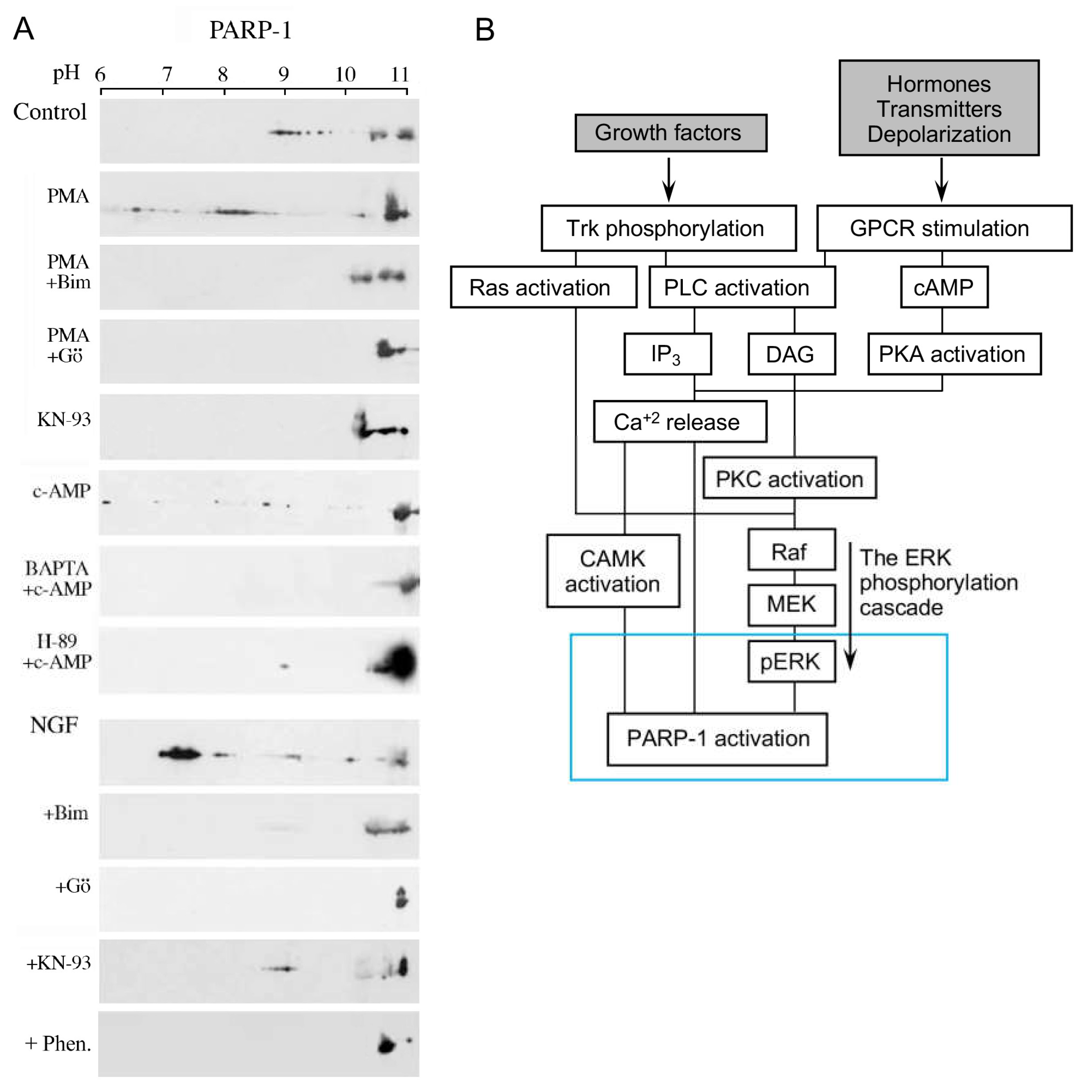
3. A Long-Lasting PARP1 Activation by Phosphorylated Erk
This entry is adapted from the peer-reviewed paper 10.3390/cells11091576
References
- Langelier, M.-F.; Eisemann, T.; Riccio, A.A.; Pascal, J.M. PARP family enzymes:regulation and catalysis of the poly(ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 2018, 53, 187–198.
- Kallberg, T.; Langelier, M.-F.; Pascal, J.; Schuler, H. Structural biology of writers, readers, and erasers in mono- and poly(ADP-ribose)mediated signaling. Mol. Asp. Med. 2013, 34, 1089–1108.
- Dantzer, F.; Amé, J.C.; Schreiber, V.; Nakamura, J.; Ménissier-de Murcia, J.; de Murcia, G. Poly(ADP-ribose) polymerase-1 activation during DNA damage and repair. Methods Enzymol. 2006, 409, 493–510.
- Luscher, B.; Butepage, M.; Eckei, L.; Kreig, S.; Verheugd, P. ADP ribosylation, a multifaceted posttranslational modification involved in the control of cell physiology in health and disease. Chem. Rev. 2018, 118, 1092–1136.
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732.
- Poirier, G.G.; deMurcia, G.; Jongstra-Bilen, J.; Mandel, P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. USA 1982, 79, 3423–3427.
- Tulin, A.; Spradling, A. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puf loci. Science 2003, 299, 560–562.
- Kraus, W.L.; Lis, J.T. PARP goes transcription. Cell 2003, 113, 677–683.
- Krishnakumar, R.; Gamble, M.J.; Frizzell, K.M.; Berocal, J.G.; Kininis, M.; Kraus, W.L. Reciprocal binding of PARP-1 and histone H1 at promoters specifes transcriptional outcomes. Science 2008, 319, 819–821.
- Visochek, L.; Grigoryan, G.; Kalal, A.; Milshtein-Parush, H.; Gazit, N.; Slutsky, I.; Yeheskel, A.; Shainberg, A.; Castiel, A.; Seger, R. A PARP1-ERK2 synergism is required for the induction of LTP L. Sci. Rep. (Nat.) 2016, 6, 24950.
- Izzo, A.; Schneider, R. The role of linker histone H1modifications in the regulation of gene expression and chromatin dynamics. Biochim. Biophys. Acta 2016, 1859, 486–495.
- Azad, G.K.; Ito, K.; Sailaja, T.; Biran, A.; Nissim-Rafinia, M.; Yamada, Y.; Brown, D.T.; Takizawa, T.; Meshorer, E. PARP1-dependent eviction of the linker histone H1mediates immediate early gene expression during neuronal activation. J. Cell. Biol. 2017, 217, 473–481.
- Fontan-Lozano, A.; Suarez-Pereira, I.; Horillo, A.; del-Pozo-Martin, Y.; Hmadch, A. Histone H1 Poly-Ribosylation Regulates the Chromatin Alterations Required for Learning Consolidation. J. Neurosci. 2010, 30, 13305–13313.
- Cohen-Armon, M.; Yeheskel, A.; Pascal, J.M. Signal-induced PARP1-Erk synergism mediates IEG expression. Signal Transduct. Target. Ther. 2019, 4, 8.
- Cohen-Armon, M.; Visochek, L.; Rozensal, D.; Kalal, A.; Geistrikh, I.; Klein, R.; Bendetz-Nezer, S.; Yao, Z.; Seger, R. DNA-Independent PARP-1 Activation by Phosphorylated ERK2 Increases Elk1 Activity: A Link to Histone Acetylation. Mol. Cell 2007, 25, 297–308.
- Visochek, l.; Steingart, R.; Vulin-Shultzman, I.; Klein, R.; Priel, E.; Gozes, I.; Cohen-Armon, M. PolyADP-ribosylation is involved in neurotrophic activity. J. Neurosci. 2005, 25, 7420–7428.
- Cohen-Armon, M. PARP-1 activation in the ERK signaling pathway. Trends Pharmacol. Sci. 2007, 28, 556–560.
- Visochek, L.; Cohen-Armon, M. PARP1-Erk synergism in proliferating cells. Oncotarget 2018, 9, 29140–29145.
- Berridge, M.J.; Irvine, R.F. Inositol phosphates and cell signaling. Nature 1989, 341, 197–204.
- Banno, Y.; Nagao, S.; Katada, T.; Nagata, K.; Ui, M.; Nozawa, Y. Stimulation by GTP-binding proteins (Gi, Go) of partially purified phospholipase C activity from human platelet membranes. Biochem. Biophys. Res. Commun. 1987, 146, 861–869.
- Exton, J.H. Signaling through phosphatidylcholine breakdown. J. Biol. Chem. 1990, 265, 1–4.
- Hennager, D.J.; Welsh, M.J.; Delise, S. Changes in either cytosolic or nucleoplasmic inositol 1,4,5-triphosphate levels can control nuclear Ca+2 concentration. J. Biol. Chem. 1995, 270, 4959–4962.
- Malviya, A.N.; Rogue, P.J. “Tell me where is calcium bred”: Clarifying the roles of nuclear calcium. Cell 1998, 92, 17–23.
- Gerasimenko, O.V.; Gerasimenko, V.J.; Tepikin, A.V.; Petersen, O.H. ATP-dependent accumulation and inositol trisphos phateor cyclic ADPribose-mediated release of Ca+2 from the nuclear envelope. Cell 1995, 80, 439–444.
- Cohen-Armon, M. PARP1 activation is required for long-term memory. In Long-Term Memory: Mechanisms, Types and Disorders. Series: Neurosci. Res. Progress, Perspectives on Cognitive Psychology; Alexandrov, A.K., Fedoseev, L.M., Eds.; NOVA Sci. Pub.: New York, NY, USA, 2012.
- Homburg, S.; Visochek, L.; Moran, N.; Dantzer, F.; Priel, E.; Asculai, E.; Schwartz, D.; Rotter, V.; Dekel, N.; Cohen-Armon, M. A Fast Signal–Induced Activation of Poly(Adp-Ribose) Polymerase: A Novel Downstream Target of Phospholipase C. J. Cell Biol. 2000, 150, 293–308.
- Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G.L.; Thoronton, J.M. Metal ions in biological catalysis: From Enzyme databases to general principles. J. Biol. Inorg. Chem. 2008, 13, 1205–12018.
- Ju, B.-G.; Schum, D.; Song, E.J.; Lee, K.-J.; Ross, D.W.; Glass, C.K.; Rosenfeld, M.G. Activating the PARP1-sensor component of the Groucho/TLE1 core pressor complex mediates a CaMKinase dependent neurogenic gene activation pathway. Cell 2004, 119, 815–829.
- Payne, M.E.; Fong, Y.L.; Ono, T.; Collbran, R.J.; Kemp, B.E.; Shoderling, T.R.; Means, A.R. Calcium/calmodulin-dependent protein kinase II. J. Biol Chem. 1988, 263, 7190–7199.
- Kuo, P. The mechanism of protein kinase C activation. Trends Neurosci. 1989, 12, 425–432.
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18.
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase:Multiple substrates regulate cellular functions. Growth Factors 2006, 24, 21–44.
- Khokhlachev, A.V.; Canagarajah, B.; Wilsbacher, J.; Robinson, M.; Atkinson, M.; Goldsmith, E.; Cobb, M. Phosphorylation of MAP kinase Erk2 promotes its homodimerization and nuclear translocation. Cell 1998, 91, 605–615.
- Casar, B.; Pinto, A.; Crespo, P. ERK dimers and Scaffold proteins, Un-expected partners for a forgotten (cytoplasmic) task. Cell Cycle 2009, 8, 7–17.
- Bhalla, U.S.; Iyengar, R. Emergent properties of networks of biological signaling pathways. Science 1999, 283, 381–387.
- Sasagawa, S.; Ozaki, Y.-I.; Fujita, K.; Kuroda, S. Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nat. Cell Biol. 2005, 7, 365–373.
- Buchwalter, G.; Gross, C.; Wasylyk, B. Ets ternary complex transcription factors. Gene 2004, 324, 1–14.
- Pouyssegur, J.; Lenormand, P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signaling. Eur. J. Biochem. 2003, 270, 3291–3299.
- Busca, R.; Pouyssegur, J.; Lenormand, P. ERK1 and ERK2 Map Kinases: Specific roles or functional redundancy. Front. Cell Dev. Biol. 2016, 4, 1–23.
- Geistrikh, I.; Visochek, L.; Klein, R.; Miller, L.; Mittelman, L.; Shainberg, A.; Cohen-Armon, M. Ca+2-induced PARP-1 activation and ANF expression are coupled events in cardiomyocytes. Biochem. J. 2011, 438, 337–347.
- Tanoue, T.; Adachi, M.; Moriguchi, T.; Nishida, E. A conserved docking motif in MAP kinases common to substrates activetors and regulators. Nat. Cell Biol. 2000, 2, 110–116.
- Jacobs, D.; Glossip, D.; Xing, H.; Muslin, A.J.; Kornfeld, K. Multiple docking sites on substrate proteins form modular system that mediates recognition by Erk MAP kinase. Gene Dev. 1999, 13, 163–175.
- Tanoue, T.; Nishida, E. Docking interactions in the mitogen-activated protein kinase cascades. Pharmacol. Ther. 2002, 2–3, 193–202.
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O. Anisotropy of fluctuation dynamics of protein with elastic network model. Biophys. J. 2001, 80, 505–515.