Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Physiology
|
Neurosciences
The broad distribution of voltage-gated potassium channels (VGKCs) in the human body makes them a critical component for the study of physiological and pathological function. Within the KCNQ family of VGKCs, these aqueous conduits serve an array of critical roles in homeostasis, especially in neural tissue.
- KCNQ channels
- neural plasticity
1. Introduction
Ion channels serve as an aqueous conduit for several nuanced cellular processes to maintain the homeostatic direction of the body. Moreover, there are over 400 genes that encode for at least one ion channel subunit [1,2]. The various mechanisms for alternative splicing make for an enormous variety of subunit combinations designed for appropriate physiological functions. Among these, the largest and most diverse group of ion channels are potassium (K+) channels [2,3]. These channels are composed of tetrameric integral membrane regions, which form an aqueous pore for K+ to permeate across the membrane. This ion serves a critical role in maintaining electrical gradients during the repolarization of action potentials and maintaining the negative resting membrane potential [3,4].
Voltage-gated potassium channels (VGKCs, also Kv) form a broad distribution of channels in the nervous system as well as other tissues. Structurally, Kv channels are also a tetramer integral membrane pore-forming alpha subunit but also contain six transmembrane segmental helices, classified as S1–S6. In addition, the S1–S4 transmembrane segmental helices compose the actual voltage sensation region, and the latter two (S5–S6) units are the actual gate of the channel, as depicted in Figure 1. The voltage sensation region (S1–S4) is supple in its ability to adapt to shifting membrane potentials by creating a conformational shift. This shift spreads through the pore-forming subunit via interactions with the S4 transmembrane segments. In addition, this segment is also protected during depolarization of the action potential (AP). This protection is due to the presence of the acidic residues on S1 and S2 transmembrane segments, which limits deterrence [3,4,5].
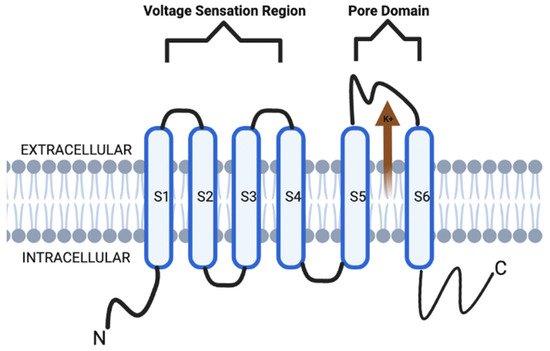
Figure 1. KCNQ channel structure is composed of six transmembrane segmental helices, classified as S1–S6. In addition, the S1–S4 transmembrane segmental helices compose the actual voltage sensation region, and the latter two (S5–S6) units are the actual gate of the channel.
Within the family of Kv channels, there are subfamilies that can be grouped according to the N- and C-terminal domains and encoded genes [5,6]. The importance behind the subfamily grouping lies in the Kv proteins, which can be functionally divergent with different membrane sensitivity potentials, gating interactions, and dynamic responses [4]. These subfamilies of Kv channels are all encoded by 40 genes, and current literature establishes exactly 12 subfamilies of Kv channels as a product of this gene encoding (e.g., Kv1–12) [6].
Historically, some of the earliest studies on voltage-gated ion channels (VGICs) were on the contemporary Kv7 subfamily [5,6]. Moreover, the understanding of the Kv7 subfamily was not immediate upon discovery. Rather, the literature initially focused on a concept known as the M channel. This channel was initially termed due to its activity as a low-threshold non-inactivating K+ channel [7]. They were named “M channels” as such because of pilot literature that showcased their inhibition via muscarinic acetylcholine receptors (mAChR) stimulation [5]. Today, the subunits of the subfamily Q Kv7 K+ (KCNQ) channel family are now known to be part of M channels and are a key target as the basis for pharmacological treatment modalities for a broad spectrum of neurological disorders. This is because Kv7 have been shown to be stimulated by membrane potentials that are more negative than the AP threshold due to their activity as a low-threshold non-inactivating K+ channel [5,6,7].
Structurally, the KCNQ channels are similar to their Kv channel relatives (Figure 1). However, the emphasis on these channels is in their ability to utilize their glycine residues to contribute to a major part of their K+ ion preference [8,9]. Specifically, the channels have glycine residues which utilize their carbonyl oxygen branches to form a shell that is specific for the size of K+ ions compared to Ca2+ and Na+ ions [9,10].
The KCNQ channels are responsible for the M currents during physiological processes, which is important in the regulation of various neuronal excitability [10]. The basis of which is formed by several different KCNQ isoforms forming heterotrimeric channels. The M-current is a non-inactivating sub-threshold current [9,10]. The increases in neuronal excitability have resulted from physiological modulation, pharmacological inhibition, and genetic mutations that affect the M-current [9,10,11]. The Kv7 channels can transiently induce the suppression of the M-current such that they limit the firing frequency of neurons [10,11]. Furthermore, it is the Kv7.2 and Kv7.3 channels which are specifically involved in the regulation of M-current, and some other channels can also play minor contributory roles [10,11,12,13].
With regards to the actual opening and closing of the KCNQ channel, there are several mechanisms. For example, KCNQ channels can open via binding of the phosphatidylinositol 4,5-bisphosphate (PIP2) ligand. The direct binding of gamma-aminobutyric acid (GABA) to the KCNQ channel can directly increase the likelihood that a KCNQ channel will open and allow K+ permeation. This mechanism seems to be GABA-specific as such a conformation has not been identified in KCNQ channels activated by other means. Secondly, inositol 1,4,5-trisphosphate (IP3)-mediated intracellular calcium signals promote PIP2 synthesis and, via calmodulin, will suppress the M-current [14,15]. In regard to neuronal KCNQ channels, their importance lies in the ability to modulate neurotransmitter release and somatic excitation in the nervous system. Robust production of PIP2 via hydrolysis agonizes four receptors in the sympathetic neurons of the superior cervical ganglion (e.g., M1, AT1, B2, and P2Y). Modulation of this system occurs via competitive or allosteric regulation of the membrane transport protein affinities for PIP2 molecules [1,5,6,7,8,9,10,11,16,17,18,19,20,21].
With this array of physiological properties found in KCNQ channels, there has been a growth in the literature on KCNQ channel property modifications for therapeutic treatment modalities, as well as the role of these channels in various pathological processes. Specifically, the alteration or loss of function (LOF) by these KCNQ (i.e., channelopathies) highlight their importance in physiological function in the body.
There are various phenotypic presentations of these channelopathies as most are due to genetic etiology amongst whichever genes are involved and the location of the channels, as depicted in Table 1 [17,18,19,20,21]. The most common genes involved in channelopathies are KCNQ1-5 (without consideration of spliced variants) [14,17,21]. KCNQ1 is most expressed in cardiac and cochlear tissue [14,22]. Specifically, cardiac KCNQ1 LOF mutations are associated with type 1 long-QT syndrome [22,23,24,25,26,27,28,29,30]. Cochlear KCNQ1 pathology involves the autosomal recessive long-QT syndrome (Jervell Lange-Nielsen syndrome), which is associated with potassium channelopathy leading to bilateral sensorineural hearing loss as well as the cardiac arrhythmia [26,27,28,29]. KCNQ2 is most expressed in the fetal cerebellum, hippocampus, and medulla [30,31]. Genetic mutation in KCNQ2 is often associated with benign familial neonatal seizures and early-onset epileptic encephalopathy [9,32,33,34,35,36,37,38,39,40,41,42,43,44,45]. KCNQ3 is also most expressed in the fetal cerebellum, hippocampus, and medulla [9,30]. In addition, KCNQ3 mutations are often associated with channelopathies in conjunction with KCNQ2 [33], but additional literature also supports KCNQ mutations in bipolar disorder [36] and various thyroid disorders [37]. Similar to KCNQ1 expression, KCNQ4 is most expressed in the cochlear hair cells but also in trigeminal ganglia [14]. This plays a key role in maintaining the K+ gradient for channel mechanosensation to carry K+ into hair cells to stimulate auditory sensation [14,38]. KCNQ4 mutations are often associated with auditory hearing loss and have therefore been a key target in developing pharmacotherapeutic options for hearing loss [39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54]. KCNQ5 is most expressed in neural tissue, including the retinal pigment epithelium [48]. However, the lack of recent literature on the profile of these encoded channel subfamilies suggests that there may be unknown channelopathies related to vision homeostasis [14,48]. The expression of these genes is more often in association with other KCNQ genes than what was separately outlined. In addition to KCNQ5, KCNQ1 and KCNQ4 are also often encoded to channels in the neuronal retina and may also have a degree of contribution to its physiological function [9,14,30]. Despite this, the importance of highlighting single gene encoding remains key to approaching neural pathophysiology [2,8,14]. Given this importance, the aim of this review is to provide an up-to-date understanding of the contemporary work of KCNQ channels in order to provide greater emphasis on KCNQ’s involvement in various pathophysiological processes distributed throughout the human body.
Table 1. Expression distribution and associated pathologies with channel genes.
| Gene | Expression Distribution | Associated Pathologies |
|---|---|---|
| KCNQ1 | Cochlea | Type 1 long QT syndrome |
| Heart | ||
| KCNQ2 | Cerebellum | Benign familial neonatal seizures |
| Hippocampus Medulla |
Early onset epileptic encephalopathy | |
| KCNQ3 | Cerebellum | Benign familial neonatal seizures |
| Hippocampus Medulla |
Early onset epileptic encephalopathy Bipolar Disorder |
|
| KCNQ4 | Cochlea Trigeminal ganglia |
Deafness |
| KCNQ5 | Retinal pigment epithelium | * |
* No major associated pathologies. Of note, this table is not comprehensive to all expression and pathological distributions of these genes.
2. Modulation of Synaptic Plasticity by KCNQ Channels
There has been a greater development in the role of KCNQ channels among neuronal networks in the past decade. This has led to its consideration for potential pharmacotherapeutic applications [14]. The ability for neuronal modification, or neural plasticity, is a key area of focus in understanding the foundations of learning and memory functions. Anatomically, the origin of the literature on neural plasticity can be further refined by discussing the concept of synaptic plasticity. This concept focuses on hippocampal formation and two principal cell types: pyramidal neurons and granular cells. Specifically, the pyramidal neurons are composed of diverse branching of dendritic neurons, which are responsible for synaptic communication with other neurons [49,50]. The morphological formation of these neurons within the hippocampus leads to the further subfield classification of pyramidal cells in what is known as Cornu Ammonus (CA), divided into CA1, CA2, and CA3 [49,50]. These regions serve an important role in localizing KCNQ channel function in synaptic plasticity [49,51,52,53,54,55,56,57,58,59,60,61]. Within synaptic plasticity, two major models involved in the application of neural plasticity are long-term potentiation (LTP) and long-term depression (LTD) [59]. These models are activity-dependent, and the literature establishes their role in namesake enhancement or reduction in synaptic efficiency. Historically, LTP was initially found in animal models, which found a sustained enhancement in the hippocampus following high-frequency electrode stimulation. LTD was later recognized after laboratory models found the opposite effect following low-frequency simulations [61,62,63,64,65]. At the cellular level, the literature suggests there are numerous factors that play a role in creating the genres of synaptic efficiency and, ultimately, neural plasticity [63,64,65,66,67,68].
Historically, the literature establishes high concentrations of KCNQ2–5 channels in the perisomatic CA1 hippocampal regions [50]. Within dendritic CA1 regions, the current generated by KCNQ channels may not serve as robust of a role as seen in pyramidal CA1 regions [14,50]. Moreover, it has been seen that modulation of KCNQ currents via linopirdine and XE991 do not create effects on synaptic excitability. Rather, it has been shown that the axonal KCNQ channels create a backpropagation into the dendritic CA1 regions [14,65,66,67,68]. This may suggest that the quantity of KCNQ channels in the dendrites does not play as robust of a role in synaptic excitability as the axonal KCNQ channels do themselves [64,65,66,67,68]. This makes axonal KCNQ channels the greater focus of study.
It was initially found that pharmacologic KCNQ channel inhibition via linopirdine reduced spike frequency adaptation (SFA) in CA1 pyramidal neurons in vitro, but only after the initial spike [49,50]. Following this initial discovery, it was also found that KCNQ channel modulation also plays a role in after hyperpolarization, which ultimately supports the notion that KCNQ channels contribute to AP [66]. In addition, muscarinic channel inhibition (i.e., KCNQ) has been shown to stimulate an array of homeostatic neuroplastic changes in synaptic efficiency. This array, in combination with KCNQ’s contribution to AP, may suggest that this array occurs at different time points, which allows for understanding that a temporal process of these neuroplastic changes occurs rather than a synced process in the hippocampus [67]. If the behavior of KCNQ channels occurs in a temporal process, this can make way for a greater understanding of the role of KCNQ via LTP and, therefore, memory development.
LTP genre can be categorized as either dependent or independent of N-methyl-D-aspartate (NMDA) receptors. Within the NMDA receptor-dependent form of LTP, it is suggested that KCNQ inhibition via XE991 stimulates the opening of NMDA receptors mediated channels during LTP by stimulating the depolarization after AP firing when performed via theta-burst stimulation [65]. This behavior may suggest that XE991 inhibition could serve a pharmacotherapeutic role in improving memory. However, the literature regarding the modulation of KCNQ channels regarding memory and LTP is still not completely understood [62,63,64,65,66,67,68]. With regard to LTP in the presence of acute stress, it is well understood that stress impairs spatial memory retrieval. Flupirtine-induced activation of KCNQ channels in the CA1 region is found to have a neuroprotective effect on spatial memory retrieval in the case of acute stress. The mechanism behind these protective effects is suggested to be through the Akt/GSK-3β and Erk1/2 signaling pathways, and animal models have shown flupirtine treatments resulted in decreased expression of apoptosis factors (i.e., Bax) and upregulation of hippocampal p-Erk1/2 [66,67,68]. Likewise, literature establishes beneficial effects on memory via KCNQ pharmacological inhibition as well. In addition to the aforementioned effects on LTP by XE991, this inhibitor has also improved cognitive impairment secondary to acetylcholine (ACh) depletion animal model induced by the neuroexcitatory kainic acid [64,65,66,67,68]. The discovery of this behavior has led to a growing body of literature on potential therapeutic applications in Alzheimer’s Disease due to its nature as a cholinergic deficiency-related cognitive impairment [66,67]. In addition, the inhibition of KCNQ channels via linopirdine is also well-established in enhancing cognition via increased ACh release [63,65,66,67,68,69,70,71,72,73,74,75,76,77].
In contrast, while LTP typically occurs after a brief high-intensity stimulation of a postsynaptic neuron, LTD can be caused by prolonged low-intensity stimulation or simulation that occurs after the firing of an AP [69]. This leads to insufficient depolarization due to this lower level of stimulation. This does not generate a removal of the magnesium blockage of the NMDA receptor [78,79,80,81,82]. However, there is evidence that this stimulation is enough to open some NMDA receptors to allow for calcium ions into the cell. These cellular calcium levels are thought to activate a cellular cascade to remove α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors [77,78,79,80,81]. This reduces the postsynaptic glutamate receptor density, which decreases synapse efficiency and, therefore, memory and learning development. Despite the developments in literature dedicated to LTD, there is little literature on the effects that KCNQ channels have on this mechanism compared to LTP.
Other than the pharmacological inhibition of KCNQ channels, the inhibition by genetic proxy also serves a role in neural plasticity with regard to cognition. Animal models have shown epileptic seizures in addition to cognitive spatial memory impairment in cases of mutant or LOF genes that encode for KCNQ2 [82,83,84,85,86,87,88,89]. These effects brought upon by genetic inhibition challenge the protective and cognitive improvements seen in pharmacologic forms of KCNQ inhibition. This is where the literature ought to focus in order to determine if secondary factors that are not understood in these animal models also contribute to the memory impairment (i.e., hippocampal morphology) [90,91,92,93,94,95]. The epileptic phenotype, in conjunction with the cognitive impairment of these genetic models, may suggest additional psychomotor exploration [95,96,97,98,99,100,101,102,103,104].
This entry is adapted from the peer-reviewed paper 10.3390/membranes12050499
This entry is offline, you can click here to edit this entry!