SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) is a single-stranded RNA virus, infectious agent of coronavirus disease 2019 (COVID-19). COVID-19 is a real challenge for the protective immunity. Some people do not respond to vaccination by acquiring an appropriate immunological memory. The risk groups for this particular infection such as the elderly and people with compromised immunity (cancer patients, pregnant women, etc.) have the most serious problems in developing an adequate immune response. Therefore, dendritic cell (DC) vaccines that are loaded ex vivo with SARS-CoV-2 antigens in the optimal conditions are promising for immunization. Lymphocyte effector cells with chimeric antigen receptor (CAR-lymphocytes) are currently used mainly as anti-tumor treatment. However, CAR-lymphocytes may be successfully employed to treat viral diseases including COVID-19. Allogenic anti-SARS-CoV-2 CAR-NK-cells may be used as an emergency treatment.
- COVID19
- CAR-lymphocytes
- dendritic cells
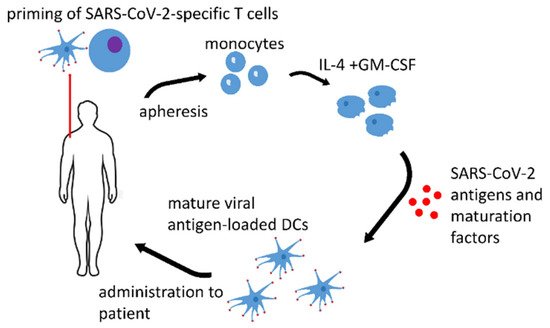
1. Dendritic Cell Vaccines
| Name of the Vaccine, ClinicalTrials.gov Identifier or Authors if Applicable, Reference | Type of the Antigen and Adjuvant |
Stage of the Study |
|---|---|---|
| AV-COVID-19, NCT04386252 [1] | S-protein with GM-CSF or without an adjuvant | Phase I–II clinical trial, not yet recruiting |
| LV-SMENP DC, NCT04276896 [2] | lentivirus vectors expressing COVID-19 minigene | Phase I–II clinical trial, recruiting |
| Zhou et al. [3] | S-protein with graphene oxide (GO) nanosheets | pre-clinical research |
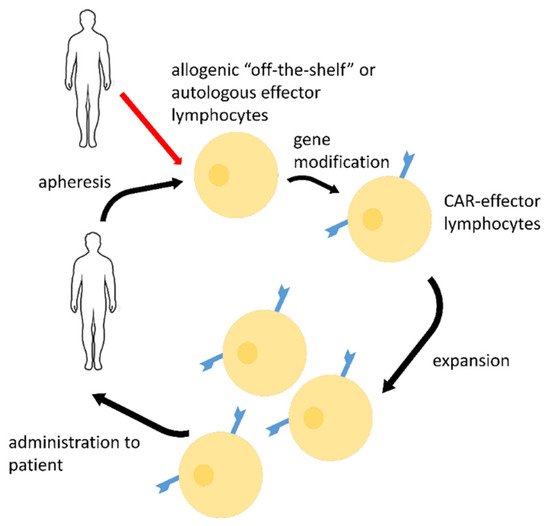
2. CAR-Effector Cell Therapy
| CAR Details, Reference | Target Antigen | Effector Cells | Stage of the Study |
|---|---|---|---|
| scFv-based CR3022 2nd generation CAR, CD28, and CD3ζ activating domains [39] |
RBD epitope | T-cells | Pre-clinical research |
| scFv-based CR3022, 3rd generation CAR, CD28, 4-1BB, and CD3ζ activating domains [48][49][50] |
RBD epitope | NK-cells | Pre-clinical research |
| scFv-based S309, 3rd generation CAR, CD28, 4-1BB, and CD3ζ activating domains [49][50] |
RBD epitope | NK-cells | Pre-clinical research |
| scFv-basedCR3022, MERTK, or MEGF10, or FcRγ(FCER1G), or CD3ζ activating domains [51] |
RBD epitope | macrophages | Pre-clinical research |
| H84T-Banana Lectin (BanLec), 2nd generation CAR, 4-1BB, and CD3 ζ activating domains [52] |
high mannose glycosites that decorate viral envelopes | NK-cells | Pre-clinical research |
| NKG2D-ACE2 CAR-NK cells for therapy of COVID-19, NCT04324996 [53] | S | NK-cells | Phase I/II clinical trial |
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10040868
References
- Phase I–II Trial of Dendritic Cell Vaccine to Prevent COVID-19 in Adults. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04386252 (accessed on 12 February 2022).
- Immunity and Safety of COVID-19 Synthetic Minigene Vaccine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04276896?term=DC&cond=SARS+CoV+2+Infection&draw=2&rank=1 (accessed on 12 February 2022).
- Zhou, Q.; Gu, H.; Sun, S.; Zhang, Y.; Hou, Y.; Li, C.; Zhao, Y.; Ma, P.; Lv, L.; Aji, S.; et al. Large-Sized Graphene Oxide Nanosheets Increase DC-T-Cell Synaptic Contact and the Efficacy of DC Vaccines against SARS-CoV-2. Adv. Mater. 2021, 33, e2102528.
- Saadeldin, M.K.; Abdel-Aziz, A.K.; Abdellatif, A. Dendritic cell vaccine immunotherapy; the beginning of the end of cancer and COVID-19. A hypothesis. Med. Hypotheses 2021, 146, 110365.
- Reuter, T.; Heldmann, M.; Schimmer, S.; Schepers, K.; Dittmer, U. Protection of mice against Friend retrovirus infection by vaccination with antigen-loaded, spleen-derived dendritic cells. Vaccine 2004, 22, 2686–2689.
- Norton, T.D.; Miller, E.A. Recent Advances in Lentiviral Vaccines for HIV-1 Infection. Front. Immunol. 2016, 7, 243.
- Mohamed, H.; Miller, V.; Jennings, S.R.; Wigdahl, B.; Krebs, F.C. The Evolution of Dendritic Cell Immunotherapy against HIV-1 Infection: Improvements and Outlook. J. Immunol. Res. 2020, 2020, 9470102.
- Norton, T.D.; Zhen, A.; Tada, T.; Kim, J.; Kitchen, S.; Landau, N.R. Lentiviral Vector-Based Dendritic Cell Vaccine Suppresses HIV Replication in Humanized Mice. Mol. Ther. 2019, 27, 960–973.
- Miller, E.; Spadaccia, M.; Sabado, R.; Chertova, E.; Bess, J.; Trubey, C.M.; Holman, R.M.; Salazar, A.; Lifson, J.; Bhardwaj, N. Autologous aldrithiol-2-inactivated HIV-1 combined with polyinosinic-polycytidylic acid-poly-L-lysine carboxymethylcellulose as a vaccine platform for therapeutic dendritic cell immunotherapy. Vaccine 2015, 33, 388–395.
- Hong, B.; Lee, S.H.; Song, X.T.; Jones, L.; Machida, K.; Huang, X.F.; Chen, S.Y. A super TLR agonist to improve efficacy of dendritic cell vaccine in induction of anti-HCV immunity. PLoS ONE 2012, 7, e48614.
- Zhou, Y.; Zhao, F.; Chen, L.; Ma, L.; Wang, Y.; He, Y.; Ma, Z.; Liu, H.; Guo, Y.; Zhang, Y.; et al. Development of a dendritic cell vaccine encoding multiple cytotoxic T lymphocyte epitopes targeting hepatitis C virus. Int. J. Mol. Med. 2013, 32, 901–909.
- Mekonnen, Z.A.; Masavuli, M.G.; Yu, W.; Gummow, J.; Whelan, D.M.; Al-Delfi, Z.; Torresi, J.; Gowans, E.J.; Grubor-Bauk, B. Enhanced T Cell Responses Induced by a Necrotic Dendritic Cell Vaccine, Expressing HCV NS3. Front. Microbiol. 2020, 11, 559105.
- Ostanin, A.A.; Chernykh, E.R. Autologous Dendritic Cell Vaccine for Treatment of Patients with Chronic HCV-Infection. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03119025?term=DC+vaccine&cond=Hepatitis+C&draw=2&rank=1 (accessed on 15 February 2022).
- Phase I–II Vaccination of Autologous Dendritic Cells Transduced with Adenoviral Vector Encoding NS3 in Hepatitis C Encoding NS3 in Hepatitis C. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT02309086?term=DC+vaccine&cond=Hepatitis+C&draw=2&rank=2 (accessed on 15 February 2022).
- Chen, M.; Li, Y.G.; Zhang, D.Z.; Wang, Z.Y.; Zeng, W.Q.; Shi, X.F.; Guo, Y.; Guo, S.H.; Ren, H. Therapeutic effect of autologous dendritic cell vaccine on patients with chronic hepatitis B: A clinical study. World J. Gastroenterol. 2005, 11, 1806–1808.
- A Clinical Trial on Hepatitis B Vaccine Activated-Dendritic Cells Combined with Anti-HBV Drugs in CHB (CTHBVACADCHB). ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02615639?term=DC+vaccine&cond=Hepatitis+B&draw=2&rank=1 (accessed on 17 February 2022).
- Luo, J.; Li, J.; Chen, R.L.; Nie, L.; Huang, J.; Liu, Z.W.; Luo, L.; Yan, X.J. Autologus dendritic cell vaccine for chronic hepatitis B carriers: A pilot, open label, clinical trial in human volunteers. Vaccine 2010, 28, 2497–2504.
- Akbar, S.M.; Furukawa, S.; Horiike, N.; Abe, M.; Hiasa, Y.; Onji, M. Safety and immunogenicity of hepatitis B surface antigen-pulsed dendritic cells in patients with chronic hepatitis B. J. Viral Hepat. 2011, 18, 408–414.
- Wei, M.J.; Pan, X.N.; Wei, K.P.; Li, X.H.; Liu, X.L.; Zhang, X.M.; Jiang, Y.L.; Zhang, C.Y.; Shen, J.K. Efficacy of HBV-pulsed DCs in combination with entecavir in patients with chronic hepatitis B infection. Int. Immunopharmacol. 2015, 27, 238–243.
- Yang, J.Y.; Cao, D.Y.; Liu, W.C.; Zhang, H.M.; Teng, Z.H.; Ren, J. Dendritic cell generated from CD34+ hematopoietic progenitors can be transfected with adenovirus containing gene of HBsAg and induce antigen-specific cytotoxic T cell responses. Cell Immunol. 2006, 240, 14–21.
- Long, J.; Zhou, B.; Li, H.; Dai, Q.; Zhang, B.; Xing, S.; Zeng, Z.; Chen, W.; Yang, J. Improvement of HBsAg gene-modified dendritic cell-based vaccine efficacy by optimizing immunization method or the application of β-glucosylceramide. Immunol. Investig. 2013, 42, 137–155.
- Chemaly, R.F.; Ullmann, A.J.; Stoelben, S.; Richard, M.P.; Bornhäuser, M.; Groth, C.; Einsele, H.; Silverman, M.; Mullane, K.M.; Brown, J.; et al. Letermovir for cytomegalovirus prophylaxis in hematopoietic-cell transplantation. N. Engl. J. Med. 2014, 370, 1781–1789.
- Van Craenenbroeck, A.H.; Smits, E.L.; Anguille, S.; Van de Velde, A.; Stein, B.; Braeckman, T.; Van Camp, K.; Nijs, G.; Ieven, M.; Goossens, H.; et al. Induction of cytomegalovirus-specific T cell responses in healthy volunteers and allogeneic stem cell recipients using vaccination with messenger RNA-transfected dendritic cells. Transplantation 2015, 99, 120–127.
- Ma, C.K.K.; Clancy, L.; Simms, R.; Burgess, J.; Deo, S.; Blyth, E.; Micklethwaite, K.P.; Gottlieb, D.J. Adjuvant Peptide Pulsed. Adjuvant Peptide Pulsed Dendritic Cell Vaccination in Addition to T Cell Adoptive Immunotherapy for Cytomegalovirus Infection in Allogeneic Hematopoietic Stem Cell Transplantation Recipients. Biol. Blood Marrow Transplant. 2018, 24, 71–77.
- Cytomegalovirus (CMV) RNA-Pulsed Dendritic Cells for Pediatric Patients and Young Adults with WHO Grade IV Glioma, Recurrent Malignant Glioma, or Recurrent Medulloblastoma (ATTAC-P). ClinicalTryals.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03615404?term=DC&cond=CMV&draw=2&rank=1 (accessed on 16 February 2022).
- Ueno, K.; Kinjo, Y.; Okubo, Y.; Aki, K.; Urai, M.; Kaneko, Y.; Shimizu, K.; Wang, D.N.; Okawara, A.; Nara, T.; et al. Dendritic cell-based immunization ameliorates pulmonary infection with highly virulent Cryptococcus gattii. Infect. Immun. 2015, 83, 1577–1586.
- Ueno, K.; Urai, M.; Ohkouchi, K.; Miyazaki, Y.; Kinjo, Y. Dendritic Cell-Based Vaccine Against Fungal Infection. Methods Mol. Biol. 2016, 1403, 537–549.
- Ueno, K.; Urai, M.; Takatsuka, S.; Abe, M.; Miyazaki, Y.; Kinjo, Y. Immunization with Antigen-Pulsed Dendritic Cells Against Highly Virulent Cryptococcus gattii Infection: Analysis of Cytokine-Producing T Cells. Methods Mol. Biol. 2017, 1625, 327–339.
- Silva, L.B.R.; Dias, L.S.; Rittner, G.M.G.; Muñoz, J.E.; Souza, A.C.O.; Nosanchuk, J.D.; Travassos, L.R.; Taborda, C.P. Dendritic Cells Primed with Paracoccidioides brasiliensis Peptide P10 Are Therapeutic in Immunosuppressed Mice with Paracoccidioidomycosis. Front. Microbiol. 2017, 8, 1057.
- Brusko, M.A.; Stewart, J.M.; Posgai, A.L.; Wasserfall, C.H.; Atkinson, M.A.; Brusko, T.M.; Keselowsky, B.G. Immunomodulatory Dual-Sized Microparticle System Conditions Human Antigen Presenting Cells into a Tolerogenic Phenotype In Vitro and Inhibits Type 1 Diabetes-Specific Autoreactive T Cell Responses. Front. Immunol. 2020, 11, 574447.
- Adorini, L.; Penna, G.; Giarratana, N.; Uskokovic, M. Tolerogenic dendritic cells induced by vitamin D receptor ligands enhance regulatory T cells inhibiting allograft rejection and autoimmune diseases. J. Cell. Biochem. 2003, 88, 227–233.
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15.
- Premkumar, L.; Segovia-Chumbez, B.; Jadi, R.; Martinez, D.R.; Raut, R.; Markmann, A.; Cornaby, C.; Bartelt, L.; Weiss, S.; Park, Y.; et al. The receptor binding domain of the viral spike protein is an immunodominant and highly specific target of antibodies in SARS-CoV-2 patients. Sci. Immunol. 2020, 5, eabc8413.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8.
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569.
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5.
- Cao, Y.; Su, B.; Guo, X.; Sun, W.; Deng, Y.; Bao, L.; Zhu, Q.; Zhang, X.; Zheng, Y.; Geng, C.; et al. Potent Neutralizing Antibodies against SARS-CoV-2 Identified by High-Throughput Single-Cell Sequencing of Convalescent Patients’ B Cells. Cell 2020, 182, 73–84.e16.
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124.
- Guo, X.; Kazanova, A.; Thurmond, S.; Saragovi, H.U.; Rudd, C.E. Effective chimeric antigen receptor T cells against SARS-CoV-2. iScience 2021, 24, 103295.
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y.; et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 2020, 9, 382–385.
- Mehrabadi, A.Z.; Ranjbar, R.; Farzanehpour, M.; Shahriary, A.; Dorostkar, R.; Hamidinejad, M.A.; Ghaleh, H.E.G. Therapeutic potential of CAR T cell in malignancies: A scoping review. Biomed. Pharm. 2022, 146, 112512.
- Björkström, N.K.; Strunz, B.; Ljunggren, H.G. Natural killer cells in antiviral immunity. Nat. Rev. Immunol. 2022, 22, 112–123.
- Carlsten, M.; Childs, R.W. Genetic manipulations of NK cells for cancer immunotherapy. Front. Immunol. 2015, 6, 266.
- Simonetta, F.; Alvarez, M.; Negrin, R.S. Natural Killer Cells in Graft-versus-Host-Disease after Allogeneic Hematopoietic Cell Transplantation. Front. Immunol. 2017, 8, 465.
- Shah, N.; Li, L.; McCarty, J.; Kaur, I.; Yvon, E.; Shaim, H.; Muftuoglu, M.; Liu, E.; Orlowski, R.Z.; Cooper, L.; et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br. J. Haematol. 2017, 177, 457–466.
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current Perspectives on “Off-The-Shelf” Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021, 12, 732135.
- Mo, F.; Mamonkin, M.; Brenner, M.K.; Heslop, H.E. Taking T-Cell Oncotherapy Off-the-Shelf. Trends Immunol. 2021, 42, 261–272.
- Ma, M.; Badeti, S.; Geng, K.; Liu, D. Efficacy of Targeting SARS-CoV-2 by CAR-NK Cells. BioRxiv 2020, 247320.
- Ma, M.; Badeti, S.; Chen, C.H.; Pinter, A.; Jiang, Q.; Shi, L.; Zhou, R.; Xu, H.; Li, Q.; Gause, W.; et al. CAR-NK Cells Effectively Target the D614 and G614 SARS-CoV-2-infected Cells. BioRxiv 2021, 426742.
- Ma, M.T.; Badeti, S.; Chen, C.H.; Kim, J.; Choudhary, A.; Honnen, B.; Reichman, C.; Calianese, D.; Pinter, A.; Jiang, Q.; et al. CAR-NK Cells Effectively Target SARS-CoV-2-Spike-Expressing Cell Lines In Vitro. Front. Immunol. 2021, 12, 652223.
- Fu, W.; Lei, C.; Ma, Z.; Qian, K.; Li, T.; Zhao, J.; Hu, S. CAR Macrophages for SARS-CoV-2 Immunotherapy. Front. Immunol. 2021, 12, 669103.
- Christodoulou, I.; Rahnama, R.; Ravich, J.W.; Seo, J.; Zolov, S.N.; Marple, A.N.; Markovitz, D.M.; Bonifant, C.L. Glycoprotein Targeted CAR-NK Cells for the Treatment of SARS-CoV-2 Infection. Front. Immunol. 2021, 12, 763460.
- A Phase I/II Study of Universal Off-the-Shelf NKG2D-ACE2 CAR-NK Cells for Therapy of COVID-19. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04324996?term=CAR&cond=COVID-19&draw=2&rank=1 (accessed on 19 February 2022).
- Pinto, D.; Park, Y.J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; De Marco, A.; et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295.
- Sohail, A.; Yu, Z.; Arif, R.; Nutini, A.; Nofal, T.A. Piecewise differentiation of the fractional order CAR-T cells-SARS-2 virus model. Results Phys. 2022, 33, 105046.
- Al-Utaibi, K.A.; Nutini, A.; Sohail, A.; Arif, R.; Tunc, S.; Sait, S.M. Forecasting the action of CAR-T cells against SARS-corona virus-II infection with branching process. Model. Earth Syst. Environ. 2021, 15, 1–9.
- Zhu, T.; Xiao, Y.; Meng, X.; Tang, L.; Li, B.; Zhao, Z.; Tan, Q.; Shan, H.; Liu, L.; Huang, X. Nanovesicles derived from bispecific CAR-T cells targeting the spike protein of SARS-CoV-2 for treating COVID-19. J. Nanobiotechnology 2021, 19, 391.
- Bednar, C.; Ensser, A. CARs-A New Perspective to HCMV Treatment. Viruses 2021, 13, 1563.
- Seif, M.; Einsele, H.; Löffler, J. CAR T Cells Beyond Cancer: Hope for Immunomodulatory Therapy of Infectious Diseases. Front. Immunol. 2019, 10, 2711.
- Slabik, C.; Kalbarczyk, M.; Danisch, S.; Zeidler, R.; Klawonn, F.; Volk, V.; Krönke, N.; Feuerhake, F.; Ferreira de Figueiredo, C.; Blasczyk, R.; et al. CAR-T Cells Targeting Epstein-Barr Virus gp350 Validated in a Humanized Mouse Model of EBV Infection and Lymphoproliferative Disease. Mol. Ther. Oncolytics 2020, 18, 504–524.
- Tang, X.; Zhou, Y.; Li, W.; Tang, Q.; Chen, R.; Zhu, J.; Feng, Z. T cells expressing a LMP1-specific chimeric antigen receptor mediate antitumor effects against LMP1-positive nasopharyngeal carcinoma cells in vitro and in vivo. J. Biomed. Res. 2014, 28, 468–475.
- Kieser, A.; Sterz, K.R. The Latent Membrane Protein 1 (LMP1). Curr. Top. Microbiol. Immunol. 2015, 391, 119–149.
- LMP1 Positive Infectious Diseases and Hematological Malignancies. ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04657965?term=CAR&cond=Infections&draw=2&rank=3 (accessed on 23 February 2022).
- Maldini, C.R.; Ellis, G.I.; Riley, J.L. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 2018, 18, 605–616.
- Liu, L.; Patel, B.; Ghanem, M.H.; Bundoc, V.; Zheng, Z.; Morgan, R.A.; Rosenberg, S.A.; Dey, B.; Berger, E.A. Novel CD4-Based Bispecific Chimeric Antigen Receptor Designed for Enhanced Anti-HIV Potency and Absence of HIV Entry Receptor Activity. J. Virol. 2015, 89, 6685–6694.
- Zhen, A.; Peterson, C.W.; Carrillo, M.A.; Reddy, S.S.; Youn, C.S.; Lam, B.B.; Chang, N.Y.; Martin, H.A.; Rick, J.W.; Kim, J.; et al. Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathog. 2017, 13, e1006753.
- Jiang, Z.; Liang, H.; Pan, H.; Liang, Y.; Wang, H.; Yang, X.; Lu, P.; Zhang, X.; Yang, J.; Zhang, D.; et al. HIV-1-Specific CAR-T Cells With Cell-Intrinsic PD-1 Checkpoint Blockade Enhance Anti-HIV Efficacy in vivo. Front. Microbiol. 2021, 12, 684016.
- Pampusch, M.S.; Abdelaal, H.M.; Cartwright, E.K.; Molden, J.S.; Davey, B.C.; Sauve, J.D.; Sevcik, E.N.; Rendahl, A.K.; Rakasz, E.G.; Connick, E.; et al. CAR/CXCR5-T cell immunotherapy is safe and potentially efficacious in promoting sustained remission of SIV infection. PLoS Pathog. 2022, 18, e1009831.
- Leslie, G.J.; Wang, J.; Richardson, M.W.; Haggarty, B.S.; Hua, K.L.; Duong, J.; Secreto, A.J.; Jordon, A.P.; Romano, J.; Kumar, K.E.; et al. Potent and Broad Inhibition of HIV-1 by a Peptide from the gp41 Heptad Repeat-2 Domain Conjugated to the CXCR4 Amino Terminus. PLoS Pathog. 2016, 12, e1005983.
- Maldini, C.R.; Gayout, K.; Leibman, R.S.; Dopkin, D.L.; Mills, J.P.; Shan, X.; Glover, J.A.; Riley, J.L. HIV-Resistant and HIV-Specific CAR-Modified CD4+ T Cells Mitigate HIV Disease Progression and Confer CD4+ T Cell Help In Vivo. Mol. Ther. 2020, 28, 1585–1599.
- Kim, G.B.; Hege, K.; Riley, J.L. CAR Talk: How Cancer-Specific CAR T Cells Can Instruct How to Build CAR T Cells to Cure HIV. Front. Immunol. 2019, 10, 2310.
- CD4 CAR+ ZFN-Modified T Cells in HIV Therapy. ClinicalTryal.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03617198 (accessed on 23 February 2022).
- CAR-T Cells for HIV Infection. ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04648046?term=CAR&cond=Infections&draw=2&rank=1 (accessed on 23 February 2022).
- Third-Generation CAR-T-Cell Therapy in Individuals with HIV-1 Infection (TCTIWHI). ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04863066?term=CAR&cond=Infections&draw=2&rank=2 (accessed on 23 February 2022).
- The Effect of Chimeric Antigen Receptor (CAR)-T Cell Therapy on the Reconstitution of HIV-specific Immune Function. ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03240328?term=CAR&cond=Infections&draw=2&rank=10 (accessed on 23 February 2022).
- Meng, Z.; Chen, Y.; Lu, M. Advances in Targeting the Innate and Adaptive Immune Systems to Cure Chronic Hepatitis B Virus Infection. Front. Immunol. 2020, 10, 3127.
- Bohne, F.; Chmielewski, M.; Ebert, G.; Wiegmann, K.; Kürschner, T.; Schulze, A.; Urban, S.; Krönke, M.; Abken, H.; Protzer, U. T cells redirected against hepatitis B virus surface proteins eliminate infected hepatocytes. Gastroenterology 2008, 134, 239–247.
- Krebs, K.; Böttinger, N.; Huang, L.R.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jäger, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology 2013, 145, 456–465.
- Kruse, R.L.; Shum, T.; Tashiro, H.; Barzi, M.; Yi, Z.; Whitten-Bauer, C.; Legras, X.; Bissig-Choisat, B.; Garaigorta, U.; Gottschalk, S.; et al. HBsAg-redirected T cells exhibit antiviral activity in HBV-infected human liver chimeric mice. Cytotherapy 2018, 20, 697–705.
- Klopp, A.; Schreiber, S.; Kosinska, A.D.; Pulé, M.; Protzer, U.; Wisskirchen, K. Depletion of T cells via Inducible Caspase 9 Increases Safety of Adoptive T-Cell Therapy Against Chronic Hepatitis B. Front. Immunol. 2021, 12, 734246.
- Festag, M.M.; Festag, J.; Fräßle, S.P.; Asen, T.; Sacherl, J.; Schreiber, S.; Mück-Häusl, M.A.; Busch, D.H.; Wisskirchen, K.; Protzer, U. Evaluation of a Fully Human, Hepatitis B Virus-Specific Chimeric Antigen Receptor in an Immunocompetent Mouse Model. Mol. Ther. 2019, 27, 947–959.
- Sautto, G.A.; Wisskirchen, K.; Clementi, N.; Castelli, M.; Diotti, R.A.; Graf, J.; Clementi, M.; Burioni, R.; Protzer, U.; Mancini, N. Chimeric antigen receptor (CAR)-engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut 2016, 65, 512–523.