Pharmacogenetics (PGx) is an emerging field of pharmacology focusing on how gene variations affect the patient’s response to treatment. Pharmacogenetics is a promising tool to optimize the selection and dosing of medications, including urate-lowering therapies (ULTs) among patients with gout. The global prevalence of gout is rising, and it disproportionately affects specific racial groups and individuals with select socioeconomic status. Genetic and experimental findings have provided evidence that genetic polymorphisms associated with serum urate pathology are also of pharmacogenetic interest. Patients with gout present with several comorbidities, warranting the use of several acute and long-term medications that increase their pill burden and the risk of adverse drug events. Implementing PGx testing can identify individuals who are more or less likely to benefit from a given treatment, improve medication adherence, and reduce pill burden.
- gout
- pharmacogenetics
- precision medicine
- genetics
- pharmacogenomics
- urate-lowering therapy
1. Background
2. Genetics of Hyperuricemia and Gout
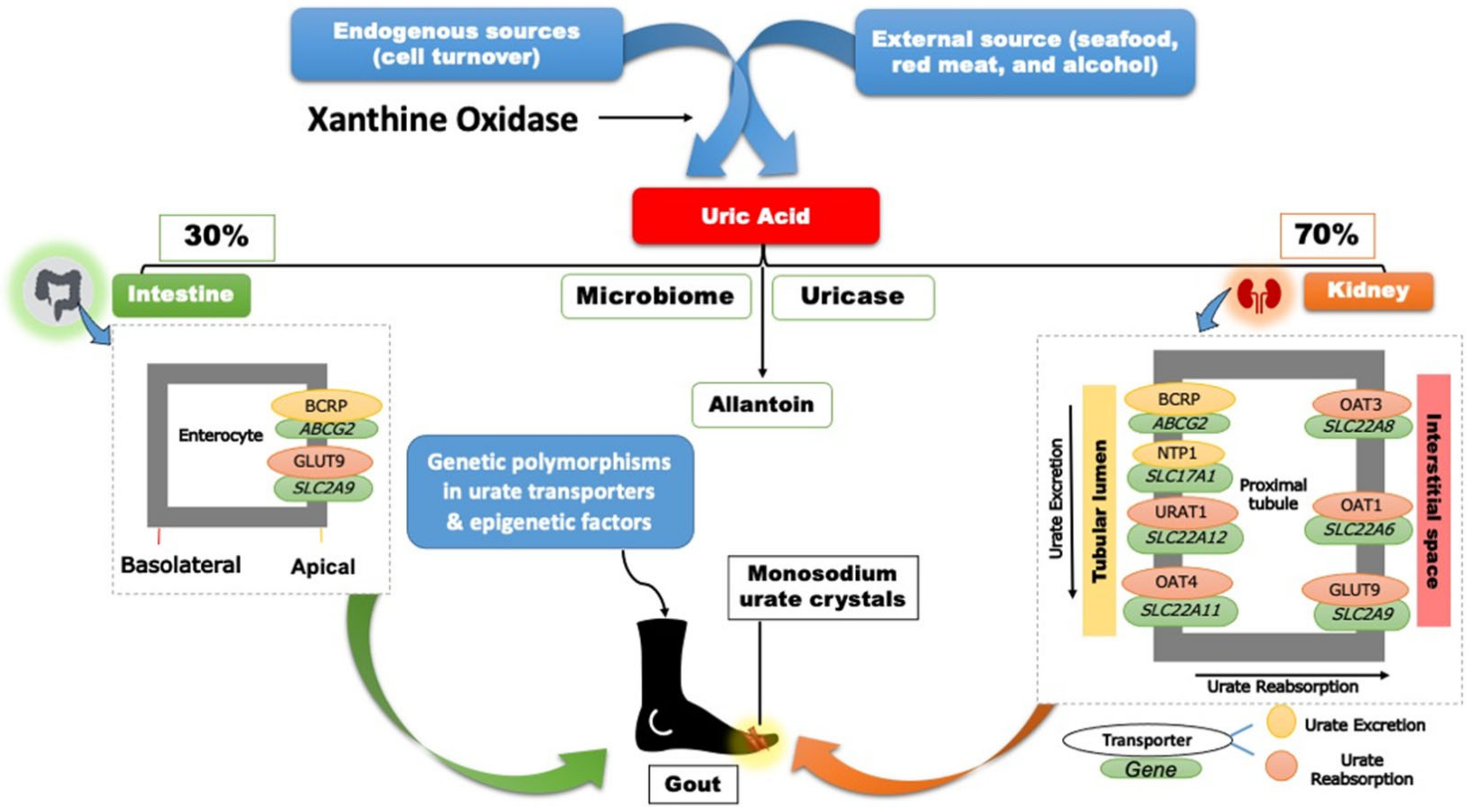
3. Gout Management Pharmacotherapy
| Drug | Mapped Genes | Effect | Clinical Outcomes | CPIC Guideline Level of Evidence a | References |
|---|---|---|---|---|---|
| Xanthine oxidase inhibitors (XO) | |||||
| Allopurinol or Oxypurinol | HLA-B | Safety | HLA-B*58:01 allele significantly increases the risk of allopurinol-induced serious cutaneous reaction | A | [2][20] |
| AOX | Response | rs3731722 A>G is associated with a better response to the standard dose of allopurinol (300 mg/day) vs. non-carriers | NA | [21] | |
| ABCG2 | Response/PK | rs2231142 C>A (Q141K) is associated with poor response to allopurinol | NA | [22] | |
| SLC22A12 | Response/PK | rs505802 C>T may influence the response to allopurinol and the PK of oxypurinol as they are substrates for the URAT1 | NA | [23][24][25] | |
| Febuxostat | UGT1A1 | Response/PK | rs34650714 C>T is associated with lower doses of febuxostat | NA | [21] |
| Uricosuric Agents | |||||
| Probenecid | SLC22A12 | Response | Homozygous or heterozygous for the mutant allele (G774A) have impaired response to loading tests of probenecid | NA | [26][27] |
| ABCB1 | PK | rs1045642 C>T could influence the PK effect of probenecid as an inhibitor when co-administered with Beta-lactam | NA | [28] | |
| G6PD | Safety | Possible hematologic adverse reactions in G6PD deficient patients | B | [29] | |
| Benzbromarone | CYP2C9 | Safety | Carriers of the no-function allele (CYP2C9*3) have reduced metabolic activity leading to prolonged exposure to benzbromarone relative to normal metabolizers | NA | [30][31] |
| Recombinant Uricase | |||||
| Pegloticase | G6PD | Safety | Risk of hemolysis or methemoglobinemia in G6PD deficient patients | B | [32] |
| Non-steroidal anti-inflammatory drugs (NSAIDs) | |||||
| Ibuprofen, celecoxib, and other NSAIDs | CYP2C9 | Safety/PK | Increased risk of NSAID-related GI bleeding in no-function allele (*3) carriers relative to normal function, as well as reduced metabolism and prolonged exposure to ibuprofen and celecoxib in CYP2C9 poor metabolizers | A (ibuprofen and celecoxib); C (indomethacin, diclofenac, naproxen) | [33][34] |
| Anti-inflammatory | |||||
| Colchicine | CYP2D6 | Response | Diminished response to colchicine in CYP2D6*4 variant carriers | NA | [35] |
| ABCB1 | Inconsistent evidence wherein one study indicates good response in the T allele carriers of the SNP rs10455642 C>T, while another study suggests no response with the T allele | NA | [36][37] | ||
| SEPHS1 | Safety | The risk allele G of rs74795203 A>G significantly increases the risk of gastrointestinal adverse events by 2.5-fold with using colchicine | NA | [38] | |
| KIF13A, RNU6-793Pb | Safety | The risk allele A of rs6916345 G>A (intergenic) was significantly associated with a ~2-fold increased risk of gastrointestinal adverse events with colchicine compared with the G allele | NA | [38] | |
| Corticosteroids | |||||
| Injectable triamcinolone acetonide | HCG22 | Safety | The G and T alleles of rs3873352 C>G and rs2523864 C>T, respectively, increase the risk of steroid-induced ocular hypertension | NA | [39] |
| IL-1 inhibitor | |||||
| Anakinra | IL1RN | Response | SNP cluster in strong linkage disequilibrium associated with poor response to anakinra | NA | [40] |
This entry is adapted from the peer-reviewed paper 10.3390/futurepharmacol2020011
References
- Roden, D.M.; McLeod, H.L.; Relling, M.V.; Williams, M.S.; Mensah, G.A.; Peterson, J.F.; van Driest, S.L. Pharmacogenomics. Lancet 2019, 394, 521–532.
- Fitzgerald, J.D.; Dalbeth, N.; Mikuls, T.; Brignardello-Petersen, R.; Guyatt, G.; Abeles, A.M.; Gelber, A.C.; Harrold, L.R.; Khanna, D.; King, C.; et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis Rheumatol. 2020, 72, 879–895.
- Murdoch, R.; Barry, M.J.; Choi, H.K.; Hernandez, D.; Johnsen, B.; Labrador, M.; Reid, S.; Singh, J.A.; Terkeltaub, R.; Mellado, J.V.; et al. Original research: Gout, Hyperuricaemia and Crystal-Associated Disease Network (G-CAN) common language definition of gout. RMD Open 2021, 7, e001623.
- Kenny, J.-E.S.; Goldfarb, D.S. Update on the Pathophysiology and Management of Uric Acid Renal Stones. Curr. Rheumatol. Rep. 2010, 12, 125–129.
- Xu, X.; Huang, J.; Wu, S.; Ji, Q.; Guo, X.; Huang, Y. The Association between the Serum Uric Acid Level and Hypertension in Middle-Aged and Elderly Adults. Cardiovasc. Ther. 2021, 2021, 4626062.
- Lanaspa, M.A.; Andres-Hernando, A.; Kuwabara, M. Uric acid and hypertension. Hypertens. Res. 2020, 43, 832–834.
- Hu, X.; Rong, S.; Wang, Q.; Sun, T.; Bao, W.; Chen, L.; Liu, L. Association between plasma uric acid and insulin resistance in type 2 diabetes: A Mendelian randomization analysis. Diabetes Res. Clin. Pract. 2021, 171, 108542.
- Hisatome, I.; Li, P.; Miake, J.; Taufiq, F.; Mahati, E.; Maharani, N.; Utami, S.B.; Kuwabara, M.; Bahrudin, U.; Ninomiya, H. Uric Acid as a Risk Factor for Chronic Kidney Disease and Cardiovascular Disease—Japanese Guideline on the Management of Asymptomatic Hyperuricemia. Circ. J. 2021, 85, 130–138.
- Wright, A.F.; Rudan, I.; Hastie, N.D.; Campbell, H. A “complexity” of urate transporters. Kidney Int. 2010, 78, 446–452.
- Yang, Q.; Kottgen, A.; Dehghan, A.; Smith, A.V.; Glazer, N.L.; Chen, M.H.; Chasman, D.I.; Aspelund, T.; Eiriksdottir, G.; Harris, T.B.; et al. Multiple Genetic Loci Influence Serum Urate Levels and Their Relationship with Gout and Cardiovascular Disease Risk Factors. Circ. Cardiovasc. Genet. 2010, 3, 523–530.
- Kolz, M.; Johnson, T.; Sanna, S.; Teumer, A.; Vitart, V.; Perola, M.; Mangino, M.; Albrecht, E.; Wallace, C.; Farrall, M.; et al. Meta-Analysis of 28,141 Individuals Identifies Common Variants within Five New Loci That Influence Uric Acid Concentrations. PLoS Genet. 2009, 5, e1000504.
- Kottgen, A.; Albrecht, E.; Teumer, A.; Vitart, V.; Krumsiek, J.; Hundertmark, C.; Pistis, G.; Ruggiero, D.; O’Seaghdha, C.M.; Haller, T.; et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 2013, 45, 145–154.
- Tin, A.; Marten, J.; Kuhns, V.L.H.; Li, Y.; Wuttke, M.; Kirsten, H.; Sieber, K.B.; Qiu, C.; Gorski, M.; German Chronic Kidney Disease Study; et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 2019, 51, 1459–1474.
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452.
- Anzai, N.; Jutabha, P.; Amonpatumrat-Takahashi, S.; Sakurai, H. Recent advances in renal urate transport: Characterization of candidate transporters indicated by genome-wide association studies. Clin. Exp. Nephrol. 2012, 16, 89–95.
- Woodward, O.M.; Köttgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Köttgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342.
- Caulfield, M.J.; Munroe, P.B.; O’Neill, D.; Witkowska, K.; Charchar, F.; Doblado, M.; Evans, S.; Eyheramendy, S.; Onipinla, A.; Howard, P.; et al. SLC2A9 Is a High-Capacity Urate Transporter in Humans. PLoS Med. 2008, 5, 1509–1523.
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.; Floyd, J.; Palmer, C.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442.
- Hung, S.-I.; Chung, W.-H.; Liou, L.-B.; Chu, C.-C.; Lin, M.; Huang, H.-P.; Lin, Y.-L.; Lan, J.-L.; Yang, L.-C.; Hong, H.-S.; et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc. Natl. Acad. Sci. USA 2005, 102, 4134–4139.
- Saito, Y.; Stamp, L.K.; Caudle, K.E.; Hershfield, M.; McDonagh, E.M.; Callaghan, J.T.; Tassaneeyakul, W.; Mushiroda, T.; Kamatani, N.; Goldspiel, B.R.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for human leukocyte antigen B (HLA-B) genotype and allopurinol dosing: 2015 update. Clin. Pharmacol. Ther. 2016, 99, 36–37.
- Carroll, M.B.; Smith, D.M.; Shaak, T.L. Genomic sequencing of uric acid metabolizing and clearing genes in relationship to xanthine oxidase inhibitor dose. Rheumatol. Int. 2017, 37, 445–453.
- Vora, B.; Brackman, D.J.; Zou, L.; Garcia-Cremades, M.; Sirota, M.; Savic, R.M.; Giacomini, K.M. Oxypurinol pharmacokinetics and pharmacodynamics in healthy volunteers: Influence of BCRP Q141K polymorphism and patient characteristics. Clin. Transl. Sci. 2021, 14, 1431–1443.
- Alghubayshi, A.; Edelman, A.; Alrajeh, K.; Roman, Y. Genetic assessment of hyperuricemia and gout in Asian, Native Hawaiian, and Pacific Islander subgroups of pregnant women: Biospecimens repository cross-sectional study. BMC Rheumatol. 2022, 6, 1.
- Iwanaga, T. Involvement of Uric Acid Transporter in Increased Renal Clearance of the Xanthine Oxidase Inhibitor Oxypurinol Induced by a Uricosuric Agent, Benzbromarone. Drug Metab. Dispos. 2005, 33, 1791–1795.
- Roman, Y.M.; Culhane-Pera, K.A.; Menk, J.; Straka, R.J. Assessment of genetic polymorphisms associated with hyperuricemia or gout in the Hmong. Pers. Med. 2016, 13, 429–440.
- Ichida, K.; Hosoyamada, M.; Hisatome, I.; Enomoto, A.; Hikita, M.; Endou, H.; Hosoya, T. Clinical and Molecular Analysis of Patients with Renal Hypouricemia in Japan-Influence of URAT1 Gene on Urinary Urate Excretion. J. Am. Soc. Nephrol. 2004, 15, 164–173.
- Hamada, T.; Ichida, K.; Hosoyamada, M.; Mizuta, E.; Yanagihara, K.; Sonoyama, K.; Sugihara, S.; Igawa, O.; Hosoya, T.; Ohtahara, A.; et al. Uricosuric Action of Losartan via the Inhibition of Urate Transporter 1 (URAT 1) in Hypertensive Patients. Am. J. Hypertens. 2008, 21, 1157–1162.
- Beringer, P.M.; Kriengkauykiat, J.; Zhang, X.; Hidayat, L.; Liu, S.; Louie, S.; Synold, T.; Burckart, G.J.; Rao, P.A.; Shapiro, B.; et al. Lack of Effect of P-glycoprotein Inhibition on Renal Clearance of Dicloxacillin in Patients with Cystic Fibrosis. Pharmacotherapy 2008, 28, 883–894.
- Chan, T.K.; Todd, D.; Tso, S.C. Drug-induced haemolysis in glucose-6-phosphate dehydrogenase deficiency. Br. Med. J. 1976, 2, 1227.
- Uchida, S.; Shimada, K.; Misaka, S.; Imai, H.; Katoh, Y.; Inui, N.; Takeuchi, K.; Ishizaki, T.; Yamada, S.; Ohashi, K.; et al. Benzbromarone Pharmacokinetics and Pharmacodynamics in Different Cytochrome P450 2C9 Genotypes. Drug Metab. Pharmacokinet. 2010, 25, 605–610.
- Dalbeth, N.; Stamp, L.K.; Merriman, T.R. The genetics of gout: Towards personalised medicine? BMC Med. 2017, 15, 108.
- Relling, M.V.; McDonagh, E.M.; Chang, T.; Caudle, K.E.; McLeod, H.L.; Haidar, C.E.; Klein, T.; Luzzatto, L. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for Rasburicase Therapy in the Context of G6PD Deficiency Genotype. Clin. Pharmacol. Ther. 2014, 96, 169–174.
- Theken, K.N.; Lee, C.R.; Gong, L.; Caudle, K.E.; Formea, C.M.; Gaedigk, A.; Klein, T.E.; Agúndez, J.A.; Grosser, T. Clinical Pharmacogenetics Implementation Consortium Guideline (CPIC) for CYP2C9 and Nonsteroidal Anti-Inflammatory Drugs. Clin. Pharmacol. Ther. 2020, 108, 191–200.
- Figueiras, A.; Estany-Gestal, A.; Aguirre, C.; Ruiz, B.; Vidal, X.; Carvajal, A.; Salado, I.; Salgado-Barreira, A.; Rodella, L.; Moretti, U.; et al. CYP2C9 variants as a risk modifier of NSAID-related gastrointestinal bleeding: A case-control study. Pharmacogenet. Genom. 2016, 26, 66–73.
- Yalcıntepe, S.; Ozdemır, O.; Sılan, C.; Ozen, F.; Uludag, A.; Candan, F.; Sılan, F. The CYP4502D6 *4 and *6 alleles are the molecular genetic markers for drug response: Implications in colchicine non-responder FMF patients. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 281–286.
- Bezalel, Y.; Gershoni-Baruch, R.; Dagan, E.; Lidar, M.; Livneh, A. The 3435T polymorphism in the ABCB1 gene and colchicine unre-sponsiveness in familial Mediterranean fever. Clin. Exp. Rheumatol. 2009, 7, S103–S104. Available online: https://www.clinexprheumatol.org/article.asp?a=3689 (accessed on 24 February 2022).
- Babaoglu, M.O.; Yasar, U.; Tufan, A.; Akdogan, A.; Calguneri, M.; Kiraz, S.; Bozkurt, A. Association of the 3435C > T polymorphism of the drug transporter gene ABCB1 with colchicine response in patients with familial Mediterranean fever. FASEB J. 2007, 21, A414–A415.
- Dubé, M.-P.; Legault, M.-A.; Lemaçon, A.; Perreault, L.-P.L.; Fouodjio, R.; Waters, D.D.; Kouz, S.; Pinto, F.J.; Maggioni, A.P.; Diaz, R.; et al. Pharmacogenomics of the Efficacy and Safety of Colchicine in COLCOT. Circ. Genom. Precis. Med. 2021, 14, 223–229.
- Jeong, S.; Patel, N.; Edlund, C.K.; Hartiala, J.; Hazelett, D.J.; Itakura, T.; Wu, P.-C.; Avery, R.L.; Davis, J.L.; Flynn, H.W.; et al. Identification of a Novel Mucin Gene HCG22 Associated with Steroid-Induced Ocular Hypertension. Investig. Opthalmol. Vis. Sci. 2015, 56, 2737–2748.
- Pardeo, M.; Rossi, M.N.; Marafon, D.P.; Sacco, E.; Bracaglia, C.; Passarelli, C.; Caiello, I.; Marucci, G.; Insalaco, A.; Perrone, C.; et al. Early Treatment and IL1RN Single-Nucleotide Polymorphisms Affect Response to Anakinra in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2021, 73, 1053–1061.
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505.