Preeclampsia is an obstetric pathology that, surprisingly, resembles the pathology of COVID-19. Both diseases are characterized by significant alterations in the renin-angiotensin system (RAS). RAS-mediated mechanisms may explain their primary clinical-pathological features, which are suggestive of an underlying microvascular dysfunction, with induction of vasculopathy, coagulopathy, and inflammation.
- COVID-19
- SARS-CoV-2
- preeclampsia
1. Introduction
2. Pathogenesis of Preeclampsia and COVID-19
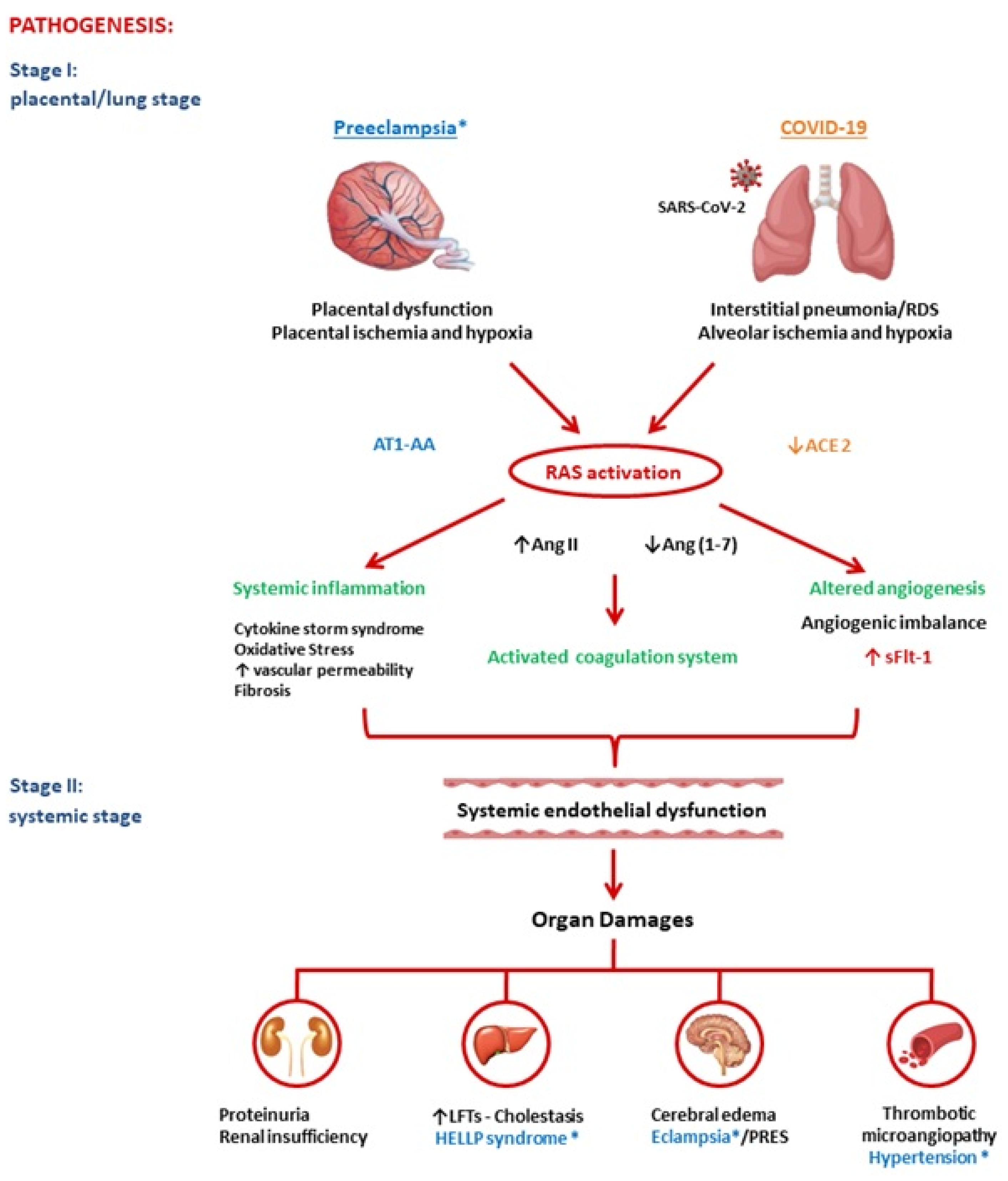
2.1. Renin-Angiotensin System
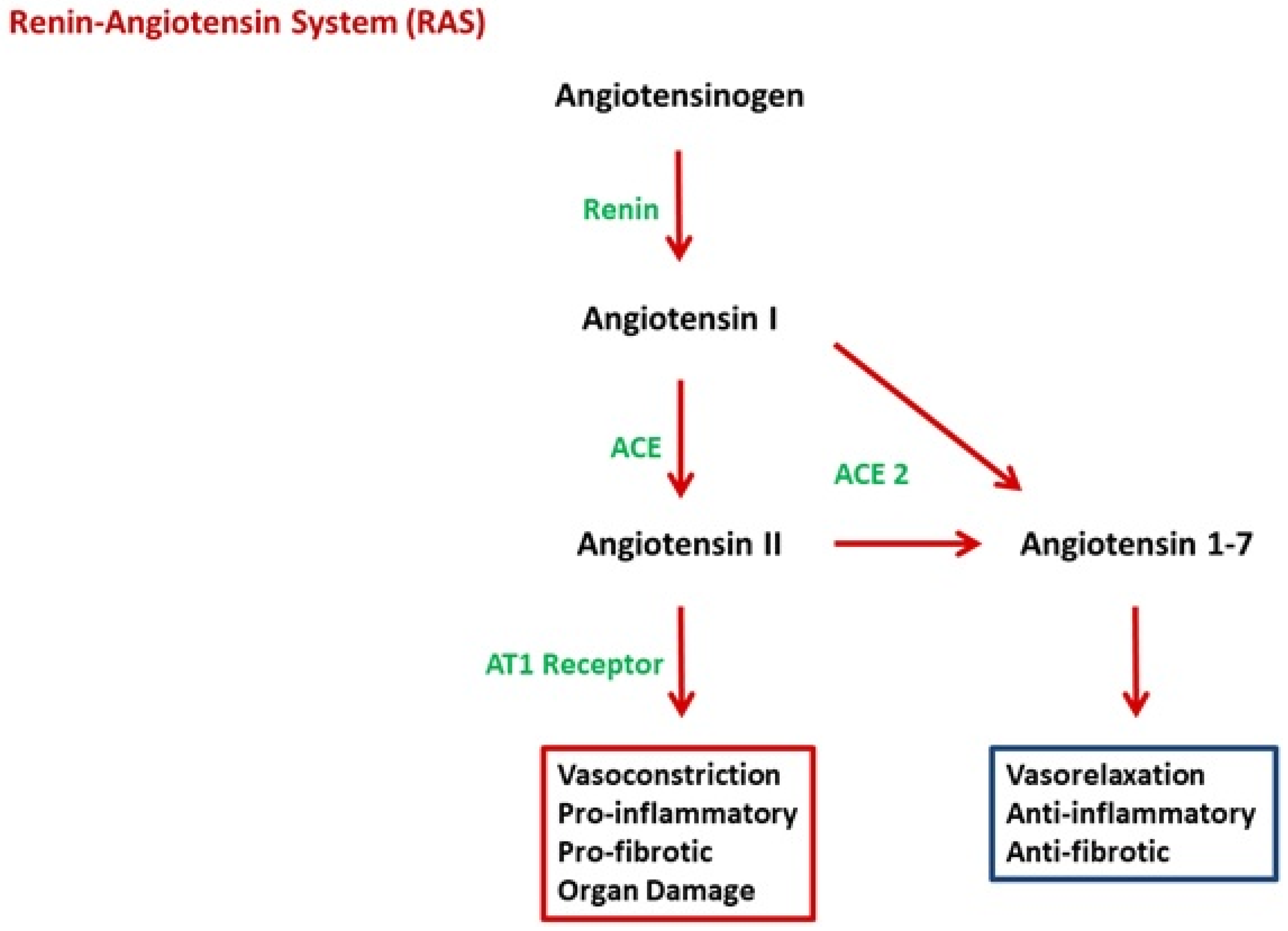
2.2. Angiotensin II
2.3. Link between RAS and sFlt-1
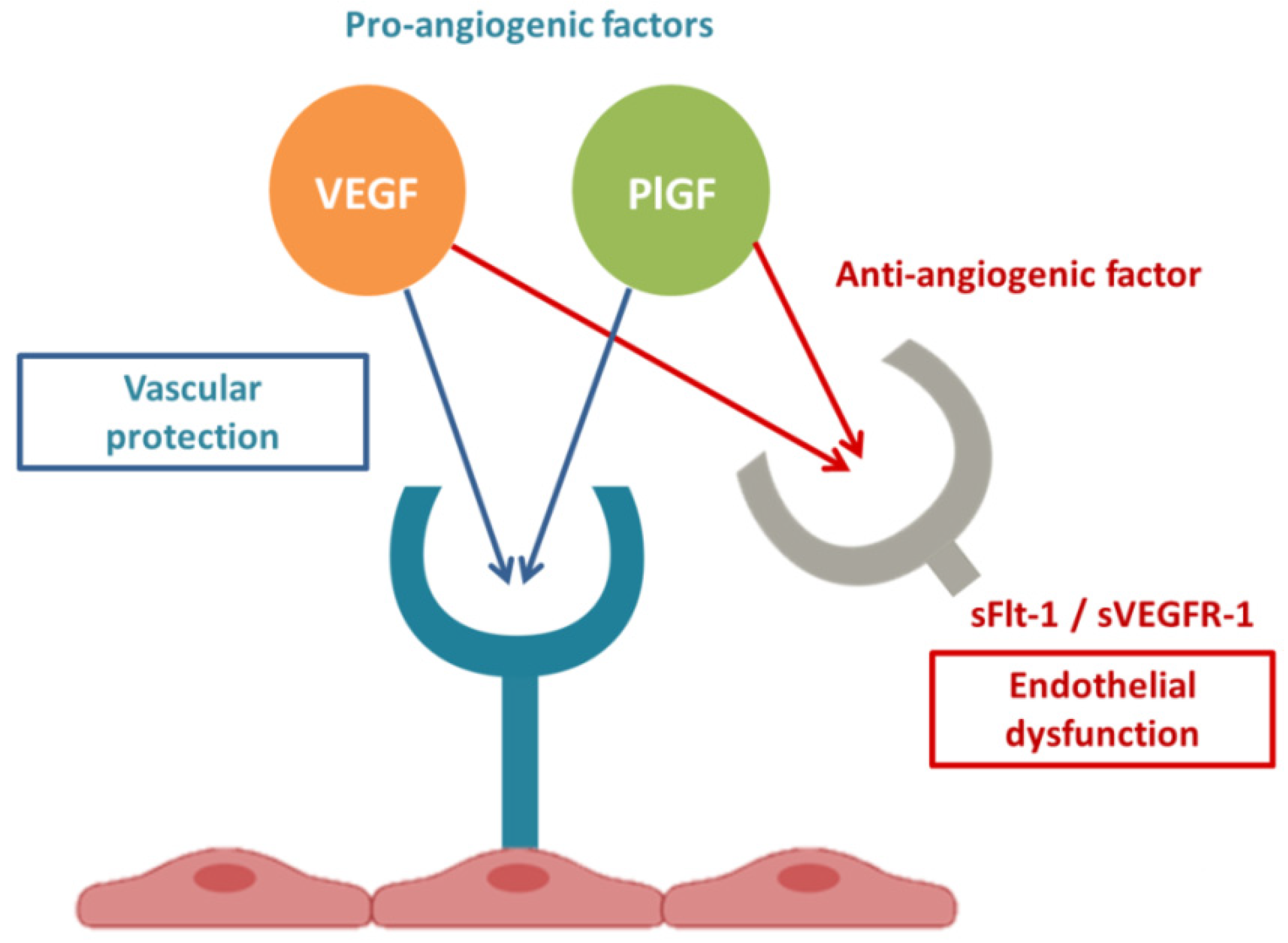
2.4. sFlt1 and Endothelial Dysfunction
3. Conclusions
PE and COVID-19 have common pathogenic pathways. Both diseases are characterized by significant alterations in the RAS with an imbalanced proportion of anti-angiogenic and pro-angiogenic soluble plasmatic factors. In summary, both PE and COVID-19 appear to be due to a state of ED secondary to increased Ang II and ensuing excessive levels of circulating anti-angiogenic factors, such as sFlt1. COVID-19 and PE are defined to be diseases that begin, respectively, in the lungs and in the placenta, and both end in the endothelium. In conclusion, SARS-CoV-2 infection could be defined an angiogenic- pneumo-syndrome.
This entry is adapted from the peer-reviewed paper 10.3390/ijtm2020016
References
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11.
- Cheng, H.; Wang, Y.; Wang, G.Q. Organ-protective effect of angiotensin-converting enzyme 2 and its effect on the prognosis of COVID-19. J. Med. Virol. 2020, 92, 726–730.
- Roberts, J.M.; August, P.A.; Bakris, G.; Barton, J.R.; Bernstein, I.M.; Druzin, M.; Gaiser, R.R.; Granger, J.P.; Jeyabalan, A.; Johnson, D.D.; et al. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131.
- Mayrink, J.; Costa, M.L.; Cecatti, J.G. Preeclampsia in 2018: Revisiting Concepts, Physiopathology, and Prediction. Sci. World J. 2018, 2018, 6268276.
- Mendoza, M.; Garcia-Ruiz, I.; Maiz, N.; Rodo, C.; Garcia-Manau, P.; Serrano, B.; Lopez-Martinez, R.M.; Balcells, J.; Fernandez-Hidalgo, N.; Carreras, E.; et al. Pre-eclampsia-like syndrome induced by severe COVID-19: A prospective observational study. BJOG 2020, 127, 1374–1380.
- Papageorghiou, A.T.; Deruelle, P.; Gunier, R.B.; Rauch, S.; García-May, P.K.; Mhatre, M.; Usman, M.A.; Abd-Elsalam, S.; Etuk, S.; Simmons, L.E.; et al. Preeclampsia and COVID-19: Results from the INTERCOVID prospective longitudinal study. Am. J. Obstet. Gynecol. 2021, 225, 289.e1–289.e17.
- Stepan, H.; Hund, M.; Andraczek, T. Combining Biomarkers to Predict Pregnancy Complications and Redefine Preeclampsia: The Angiogenic-Placental Syndrome. Hypertension 2020, 75, 918–926.
- Giardini, V.; Carrer, A.; Casati, M.; Contro, E.; Vergani, P.; Gambacorti-Passerini, C. Increased sFLT-1/PlGF ratio in COVID-19: A novel link to angiotensin II-mediated endothelial dysfunction. Am. J. Hematol. 2020, 95, E188–E191.
- Giardini, V.; Ornaghi SAcampora, E.; Vasarri, M.V.; Arienti, F.; Gambacorti-Passerini, C.; Casati, M.; Carrer, A.; Vergani, P. Letter to the Editor: SFlt-1 and PlGF Levels in Pregnancies Complicated by SARS-CoV-2 Infection. Viruses 2021, 13, 2377.
- Murphy, S.R.; Cockrell, K. Regulation of soluble fms-like tyrosine kinase-1 production in response to placental ischemia/hypoxia: Role of angiotensin II. Physiol. Rep. 2015, 3, e12310.
- Staff, A.C. The two-stage placental model of preeclampsia: An update. J. Reprod. Immunol. 2019, 134–135, 1–10.
- Lippi, G.; Sanchis-Gomar, F.; Henry, B.M. COVID-19: Unravelling the clinical progression of nature’s virtually perfect biological weapon. Ann. Transl. Med. 2020, 8, 693.
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418.
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325.
- Santos, R.A.S.; Oudit, G.Y.; Verano-Braga, T.; Canta, G.; Steckelings, U.M.; Bader, M. The renin-angiotensin system: Going beyond the classical paradigms. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H958–H970.
- Verdonk, K.; Visser, W.; Van Den Meiracker, A.H.; Danser, A.H. The renin-angiotensin-aldosterone system in pre-eclampsia: The delicate balance between good and bad. Clin. Sci. 2014, 126, 537–544.
- Walther, T.; Stepan, H. Agonist autoantibodies against the angiotensin AT1 receptor in renal and hypertensive disorders. Curr. Hypertens. Rep. 2007, 9, 128–132.
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374.
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257.
- Miesbach, W. Pathological Role of Angiotensin II in Severe COVID-19. TH Open 2020, 4, e138–e144.
- Wang, L.; Li, Y.; Qin, H.; Xing, D.; Su, J.; Hu, Z. Crosstalk between ACE2 and PLGF regulates vascular permeability during acute lung injury. Am. J. Transl. Res. 2016, 8, 1246–1252.
- Egan, K.; Kevane, B.; Ní Áinle, F. Elevated venous thromboembolism risk in preeclampsia: Molecular mechanisms and clinical impact. Biochem. Soc. Trans. 2015, 43, 696–701.
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. Global COVID-19 Thrombosis Collaborative Group, Endorsed by the ISTH, NATF, ESVM, and the IUA, Supported by the ESC Working Group on Pulmonary Circulation and Right Ventricular Function. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973.
- Ohmaru-Nakanishi, T.; Asanoma, K.; Fujikawa, M.; Fujita, Y.; Yagi, H.; Onoyama, I.; Hidaka, N.; Sonoda, K.; Kato, K. Fibrosis in Preeclamptic Placentas Is Associated with Stromal Fibroblasts Activated by the Transforming Growth Factor-β1 Signaling Pathway. Am. J. Pathol. 2018, 188, 683–695.
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140.
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; HLH Across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034.
- Irani, R.A.; Xia, Y. The functional role of the renin-angiotensin system in pregnancy and preeclampsia. Placenta 2008, 29, 763–771.
- Anton, L.; Merrill, D.C.; Neves, L.A.; Gruver, C.; Moorefield, C.; Brosnihan, K.B. Angiotensin II and angiotensin-(1-7) decrease sFlt1 release in normal but not preeclamptic chorionic villi: An in vitro study. Reprod. Biol. Endocrinol. 2010, 8, 135.
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658.
- Roberts, J.M. Objective evidence of endothelial dysfunction in preeclampsia. Am. J. Kidney Dis. 1999, 33, 992–997.
- He, Y.; Smith, S.K.; Day, K.A.; Clark, D.E.; Licence, D.R.; Charnock-Jones, D.S. Alternative splicing of vascular endothelial growth factor (VEGF)-R1 (FLT-1) pre-mRNA is important for the regulation of VEGF activity. Mol. Endocrinol. 1999, 13, 537–545.
- Shanes, E.D.; Mithal, L.B.; Otero, S.; Azad, H.A.; Miller, E.S.; Goldstein, J.A. Placental Pathology in COVID-19. Am. J. Clin. Pathol.
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennström, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Predictive Value of the sFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N. Engl. J. Med. 2016, 374, 13–22.
- Negro, A.; Fama, A.; Penna, D.; Belloni, L.; Zerbini, A.; Giuri, P.G. SFLT-1 levels in COVID-19 patients: Association with outcome
- Dupont, V.; Kanagaratnam, L.; Goury, A.; Poitevin, G.; Bard, M.; Julien, G.; Bonnivard, M.; Champenois, V.; Noel, V.; Mourvillier,
- Shapiro, N.I.; Schuetz, P.; Yano, K.; Sorasaki, M.; Parikh, S.M.; Jones, A.E.; Trzeciak, S.; Ngo, L.; Aird, W.C. The association of
- Dumnicka, P.; Sporek, M.; Mazur-Laskowska, M.; Ceranowicz, P.; Kuz ́niewski, M.; Droz ̇dz ̇, R.; Ambroz ̇y, T.; Olszanecki, R.; Kus ́nierz-Cabala, B. Serum Soluble Fms-Like Tyrosine Kinase 1 (sFlt-1) Predicts the Severity of Acute Pancreatitis. Int. J. Mol. Sci.
- Maynard, S.E.; Karumanchi, S.A. Angiogenic factors and preeclampsia. Semin. Nephrol. 2011, 31, 33–46.
- van Veen, T.R.; Panerai, R.B.; Haeri, S.; Griffioen, A.C.; Zeeman, G.G.; Belfort, M.A. Cerebral autoregulation in normal pregnancy
- van Veen, T.R.; Panerai, R.B.; Haeri, S.; Griffioen, A.C.; Zeeman, G.G.; Belfort, M.A. Cerebral autoregulation in normal pregnancy and preeclampsia. Obstet. Gynecol. 2013, 122, 1064–1069.