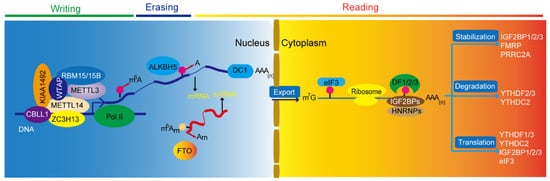
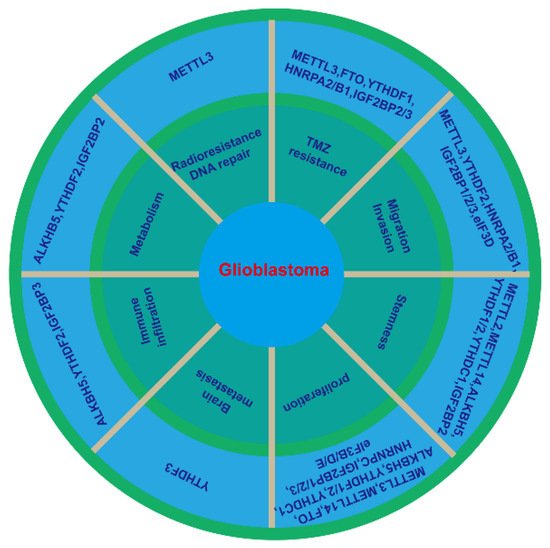
Glioblastoma is the most common and most lethal primary malignant brain tumor. N6-methyladenosine (m6A) is one of widespread and abundant internal messenger RNA (mRNA) modification found in eukaryotes. Accumulated evidence demonstrates that m6A modification is aberrantly activated in human cancers and is critical for tumorigenesis and metastasis. m6A modification is also strongly involved in key signaling pathways and is associated with prognosis in glioblastoma. Here, we briefly outline the functions of m6A and its regulatory proteins, including m6A writers, erasers, and readers on the fate of RNA. We also summarize the latest breakthroughs in this field, describe the underlying molecular mechanisms that contribute to the tumorigenesis and progression, and highlight the inhibitors targeting the factors in m6A modification in glioblastoma. Further studies focusing on the specific pathways of m6A modification could help identify biomarkers and therapeutic targets that might prevent and treat glioblastoma.
1. Introduction
Glioblastoma (CNS World Health Organization Grade 4) is a devastating type of primary brain tumor [
1], with a median survival of only 14 months, regardless of treatment. Progress has been made in understanding glioblastoma, including many genetic and immunologic profiles described in recent years [
2,
3]; however, the molecular mechanisms and epigenetic alterations that regulate and drive glioblastoma development remain largely elusive.
N
6-methyladenosine (m
6A) modification, discovered and partially characterized in a great variety of cellular mRNAs in the 1970s [
4], is the most essential and widespread internal methylation in eukaryotic mRNAs [
5]. With the development and establishment of high-throughput sequencing technologies, such as methylated RNA immunoprecipitation sequencing (MeRIP-seq/m
6A-seq) [
6], m
6A crosslinking immunoprecipitation sequencing (m
6A-CLIP-seq) [
7], m
6A individual-nucleotide-resolution crosslinking, and immunoprecipitation (miCLIP) [
8], the investigation of the m
6A RNA methylomes and mapping are over 18000 m
6A sites in the transcripts of more than 7000 human genes have been performed [
6,
9]. Methylation is a process of catalytically transferring a methyl group from an active methyl donor such as S-adenosylmethionine (SAM) to the substrate, which can chemically modify certain proteins or nucleic acids to form a methylated product [
10]. The m
6A modification has been proved to be reversible, as it involves methyltransferases (writer), demethylase (eraser), and m
6A recognized RNA binding protein (reader). m
6A regulates mRNA processing events such as alternative splicing, translation, and stability [
11] (
Figure 1). Increasing evidence shows that dysregulation of m
6A modification and its corresponding proteins are implicated in the tumorigenesis and the progression of cancers, including glioblastoma [
12,
13] (
Figure 2 and
Table 1). These studies could provide potential epigenetic targets for the diagnosis and treatment of glioblastoma.
Figure 1. m6A modification determines RNA life fate. Here, is the cycling model of methyltransferase complex, demethylase, and m6A binding proteins. A, adenosine; m6A, N6-methyladenosine; m6Am, N6, 2′-O-dimethyladenosine; Am, 2′-O-methyladenosine; snRNA, small nuclear RNA; m7G, N7-methylguanosine; AAA (n), polyadenylation.
Figure 2. m6A regulator and the hallmarks of glioblastoma. Abnormal expression of m6A writers, erasers and readers in glioblastoma affects at least eight of the hallmarks: cell proliferation, stemness, migration and invasion, TMZ resistance, radioresistance and DNA repair, metabolism, immune infiltration, and brain metastasis.
Table 1. List of reported functions of m6A regulatory proteins in glioblastoma.
| Gene Name |
Role in RNA Modification |
Role in Glioblastoma |
Mechanism |
References |
| METTL3 |
writer |
oncogene |
inhibiting sensitivity to γ-irradiation and enhancing DNA repair through recruitment of HuR to SOX2 mRNA. |
[14] |
| |
|
oncogene |
Activating NFκB in IDH-wildtype glioma after stabilization of MALAT1 |
[15] |
| |
|
oncogene |
Dysregulating the expression of epigenetically activated genes (the RNA editing, spicing and stability) |
[16] |
| |
|
oncogene |
Impairing the TMZ-sensitivity through m6A-modified DNA repair genes (MGMT and APNG)/EZH2 |
[17,18] |
| |
|
Suppressor |
Inhibiting epithelial to mesenchymal transition (EMT) and vasculogenic mimicry |
[19] |
| |
|
Suppressor |
promoting cell growth, cell differentiation, DNA damage response and cellular stress response by enhancing m6A |
[20] |
| METTL14 |
writer |
Suppressor |
promoting cell growth, cell differentiation, DNA damage response and cellular stress response by enhancing m6A |
[20] |
| WTAP |
writer |
oncogene |
A crucial interactor of the methyltransferase complex |
[21,22] |
| FTO |
eraser |
oncogene |
promoting proliferation and migration |
[23] |
| |
|
oncogene |
Increasing cell proliferation by targeting MYC transcripts |
[24] |
| |
|
Suppressor |
Inhibiting cell growth, migration and invasion by regulating m6A modification of primary pri-miR-10a processing |
[25] |
| ALKBH5 |
eraser |
oncogene |
Inhibiting cell proliferation and stemness through demethylating FOXM1 nascent transcripts and increasing HuR binding |
[26] |
| |
|
oncogene |
Demethylating G6PD transcript and enhancing its mRNA stability |
[27] |
| |
|
oncogene |
Enhancing hypoxia-induced TAM recruitment and immunosuppression by CXCL8/IL8 |
[28] |
| |
|
oncogene |
Increasing radioresistance by regulation homologous recombination |
[29] |
| |
|
oncogene |
Regulating TMZ resistance by promoting SOX2 expression |
[30] |
| YTHDF1 |
reader |
oncogene |
Promoting cell proliferation, stemness, and TMZ resistance via Musashi-1 |
[31,32] |
| YTHDF2 |
reader |
oncogene |
Positively correlating with immune cells markers, TAM markers and IDH1 |
[33] |
| |
|
oncogene |
Inhibiting cell proliferation, invasion and tumorigenesis through EGFR/SRC/ERK |
[34] |
| |
|
oncogene |
Accelerating UBXN1 mRNA degradation via METTL3-mediated m6A |
[35] |
| |
|
oncogene |
Linking epitranscriptomic modification by stabilizing MYC and VEGFA transcripts |
[36] |
| YTHDF3 |
|
oncogene |
Promoting brain metastasis through enhancing the translation of m6A-mediated transcripts (ST6GALNAC5, GJA1 and EGFR) |
[37] |
| YTHDC1 |
reader |
oncogene |
Promoting cell proliferation and stemness through VPS25-JAN-STAT |
[38,39] |
| HNRNPC |
reader |
oncogene |
Promoting cell proliferation, migration and invasion, and inhibiting apoptosis through Akt and p70S6K activation. |
[40] |
| HNRPA2/B1 |
reader |
oncogene |
Increasing cell viability, adhesion, migration, invasion, and TMZ resistance, and inhibiting apoptosis and ROS targeting STAT3, MMP-2/9 |
[41,42] |
| IGF2BP1 |
reader |
oncogene |
Targeted by non-coding RNAs and promoting cell proliferation, migration, and invasion |
[43,44,45,46,47,48] |
| IGF2BP2 |
reader |
oncogene |
Maintaining stemness and cell proliferation by regulating OXPHOS |
[49] |
| |
|
oncogene |
Targeted by non-coding RNAs and increasing TMZ resistance and proliferation |
[50,51] |
| |
|
oncogene |
Accelerating aerobic glycolysis by enhancing HK2 mRNA stability |
[52] |
| |
|
oncogene |
Promoting proliferation, and migration through recognition SRSF7 |
[53] |
| |
|
oncogene |
Promoting drug resitance by inhibition of PID1 through DANCR/FOCO1 axis |
[54] |
| IGF2BP3 |
reader |
oncogene |
Promoting proliferation, invasion and chemoresistance through PI3K and MAPK activation |
[55] |
| |
|
oncogene |
Targeted by miR-129-1 and miR-654 to induce proliferation and TMZ resistance |
[56,57] |
| |
|
oncogene |
Involving in macrophage infiltration in TME via stabilizing circNEIL3 |
[58] |
| eIF3B |
reader |
oncogene |
Promoting proliferation and inhibiting apoptosis |
[59] |
| eIF3D |
reader |
oncogene |
Promoting cell growth, colony formation and migration |
[60] |
| eIF3E |
reader |
oncogene |
Promoting proliferation through HIFs |
[61] |
2. Potential Clinical Inhibitors of m6A Modification in Glioblastoma
The writers, erasers, and readers in m6A modification are critically important for tumorigenesis and aggressiveness by regulating structure stability, RNA processing, translation, degradation, and nuclear export in glioblastoma. A novel perspective for individualized therapy of glioblastoma and other tumors may include targeting the regulators of m6A modification.
The functional domain of METTL3 that binds to the substrate of METTL3 was thought to be the target of designed inhibitors. Recently, METTL3 inhibitors, the competitive inhibitors binding the pocket of SAM, can be divided into two types: nucleosides (compounds) and non-nucleosides (UZH1A, UZH2 and STM2457). UZH1a was able to reduce m
6A/A level at least 6 days into an mRNA fraction in a leukemia cell line but also in other cell lines (U2OS, HEK293T) [
90]. The effect of METTL3 inhibitors has not been reported on glioblastoma, but we believe that it is worthy of future study.
FTO inhibitors, FTO-04, FTO-10, FTO-11, and FTO-12, reduced the size of neurospheres treated at a concentration of 30 μM. Moreover, FTO-04 can significantly impair the self-renewal of GSCs to prevent neurosphere formation without impairing the growth of human neural stem cell (hNSC) neurospheres. Importantly, treatment with FTO-04 was found to increase the levels of both m
6A and m
6Am modifications, with m
6Am modifications showing the largest fold-change relative to DMSO control. These findings make FTO-04 a leading compound for future therapy in glioblastoma [
91].
MV1035, an ALKBH5 inhibitor, can reduce U87-MG migration and invasiveness via down-regulation of CD73, which is an extrinsic protein involved in the generation of adenosine and overexpressed in glioblastoma [
92]. Under the computer-aided development of active inhibitors of ALKBH5, Simona et al. identified two low micromolar compounds among the in silico-predicted compounds by using an m
6A antibody-based enzymatic assay. These two compounds, 2-[(1-hydroxy-2-oxo-2-phenylethyl) sulfanyl] acetic acid and 4-[(furan-2-yl)-methyl] a-1,2-diazinane-3,6-dione, reduced cell viability from 100% down to about 40% at low micromolar concentrations [
93].
Lastly, a small molecule inhibitor termed 7773 interacts with a hydrophobic surface at the boundary of IGF2BP1 KH3 and KH4 domains and inhibits binding to Kras mRNA, causing a reduction in Kras mRNA and other RNA targets. This decreases cell migration and growth in cancer cell lines (H1299, ES2, and HEK293) [
94]. Kras is also upregulated in glioma samples and is involved in the ERK pathway in gliomas [
95]. Therefore, IGF2BP1 inhibitors may also be promising small molecule inhibitors for glioma treatment in the future.
3. Conclusions
Increasing evidence reveals that the writers, erasers, and readers of m6A modification play an important role in the development and tumorigenesis of glioblastoma. Moreover, some of the writers, erasers, and readers in m6A modification are potential biomarkers for diagnosis and promising drug targets for therapy in these lesions. Because glioblastoma is proven to be driven by epigenetic alteration, including mRNA manipulating the expression of oncogenes, targeting m6A regulatory proteins can serve as a new approach for precisely modifying the epitranscriptome of glioblastoma and lead to a more personalized approach to glioblastoma treatment. With the development of high-throughput sequencing technologies and chemical genetics, the studies of epitranscriptome on m6A modification have shed light on the therapy of glioblastoma. The development of inhibitors of the factors in m6A modification can control the deposition and removal of m6A marks which control RNA fate and specify the RNA to be regulated. Future efforts should be directed to the development of new inhibitors targeting the above m6A modification factors and validating their effects on glioblastoma treatment.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10050969