Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Cancer stem cells (CSCs) are known to be highly resistant to conventional therapeutic approaches, such as chemotherapeutic drugs and radiation. Therefore, selectively targeting CSCs with specific markers or signaling pathways can be an effective therapeutic strategy for treating chemotherapy-resistant liver cancer. However, there is not enough information currently available to make a conclusive statement regarding hepatic CSC-specific signaling pathways and biomarkers. In present study, we provide an overview of the current knowledge on the specific surface markers and critical signaling pathways of hepatic CSC.
- hepatic cancer stem cells
- ALDH
- CD133
- EpCAM
- Wnt/β-catenin signaling
- notch signaling
Dear author, the following contents are excerpts from your papers. They are editable.
1. Introduction
Liver cancer is the sixth most frequently diagnosed solid tumor worldwide in 2018 [1] and the third leading cause of cancer-related deaths [2]. Cancer that begins in the liver is called primary liver cancer. Hepatocellular carcinoma (HCC) represents the predominant histological subtype and accounts for approximately 80% of all primary liver cancer patients [3]. Intrahepatic cholangiocarcinoma (ICC) is the second most common primary liver cancer, representing approximately 20% of patients [4]. Both HCC and ICC are extremely heterogeneous tumors at both the genetic and phenotypic level. A newly defined mixed or combined hepatocellular carcinoma-cholangiocarcinoma (HCC-CC) characterized by dual hepatocellular and biliary epithelial differentiation suggests the existence of bipotent hepatic stem/progenitor cells with both hepatocyte and cholangiocyte lineages [5]. Indeed, recent studies indicate that HCC, ICC, and HCC-CC are highly heterogeneous in terms of their cellular and molecular characteristics and contain a small subset of self-renewing cells preferentially expressing various stem cell markers [6,7,8,9].
Furthermore, several studies have shown that purified CD133+ cells from HCC cell lines have higher proliferation potential and tumorigenic ability in animal models and exhibit stem cell-like characteristics, including their ability to self-renew and differentiate into multiple cell lineages [10]. Moreover, a subset of ICCs expresses stem/progenitor cell-related markers, suggesting CSCs are a possible cell source for ICC [11,12,13,14]. Thus, identifying and selectively targeting CSCs represents a feasible therapeutic strategy for treating liver cancer regardless of the underlying cause. However, there is not enough information currently available to make a conclusive statement regarding the cellular origin of hepatocarcinogenesis, and additional characteristics related to hepatic CSC-specific signaling pathways and markers remain to be elucidated.
2. The Origin of Cancer Stem Cells
Owing to the similarities between normal stem cells and CSCs for instance the capacity to self-renew and multi-lineage differentiation [15], many recent investigations have sought to determine whether CSCs arise from the dysregulated normal stem cells or more differentiated cells through multiple mutations. The answer may largely depend on the specific types of cancers and malignant phenotypes. The origin of CSCs is still under debate for the past few years [15,16]. Somatic stem cells are able to divide indefinitely and differentiate into some or all cell types of the tissue or organ [17]. In fact, it has been postulated that CSCs might originate from cells with stem-like characteristics or from normal stem cells by the accumulation of multiple mutations that render the stem cells cancerous [18]. Leukemic stem cells share several properties with normal hematopoietic stem cells (HSCs), supporting the stem-cell origin hypothesis [19,20]. Stem cells are usually characterized by their ability to undergo unlimited self-renewing cell division. It is therefore reasonable to hypothesize that these extended lifespan of a stem cells makes it a prime target for the multiple mutations necessary for tumor progression [21]. However, this hypothesis probably demands high mutation rates, because few somatic stem cells exist naturally in the adult tissues. Besides the stem-cell origin hypothesis, recent publications have suggested that cancer cells can also derive from fully differentiated (or “mature”) cells by undergoing de-differentiation to become more stem cell-like characteristics [22,23]. In this hypothesis, tumorigenesis is initiated by oncogenic mutations in a differentiated cell and subsequent acquisition of stem-cell-like features functions through a process of de-differentiation. Probability, the more differentiated cells exist in adult tissues, the greater the chance of mutations that can cause oncogenic transformation [24]. Surprisingly, the entire sequence of tumorigenesis can be mediated by only few steps; Takahashi et al. have recently revealed that terminally differentiated adult fibroblasts can be genetically “re-programmed” into induced pluripotent stem (iPS) cells by introducing only four transcription factors (Myc, Oct4, Sox2, and Klf4) [25,26]. However, currently there is not enough information available to make a conclusive statement regarding the origin of CSCs, and further investigation is warranted.
3. Cancer Stem Cells: Implications for Hepatocarcinogenesis
3.1. Identification of CSCs in Various Types of Tumors
The majority of cells in bulk tumors have limited tumorigenic growth and self-renewal potential; indeed, only a small population of tumor cells possess a marked self-renewal capacity, and differentiation and the ability to generate new tumors [27]. These higher tumorigenic subpopulations are known as CSCs holding a higher tumorigenic potential [28]. The CSC concept has been proposed to explain the high degree of phenotypic and functional heterogeneity of cancer cells within a given tumor [21]. In the 1960s, Bruce et al. demonstrated that only small fractions (1–4%) of leukemic cells can form colonies in in vitro and initiate new tumors in recipient animals [29]. The identification of leukemic CSCs prompted further studies to identify and isolate CSCs in various solid tumors. Extensive research in the past few decades has identified CSCs in multiple solid tumors, including colon [30], brain [31], lung [32], liver [33], and other cancers [34]. CSCs are generally defined by their distinct and specific surface antigen expression [35,36,37] and by their capacity to generate spherical colonies from single cell in suspension cultures [38]. Moreover, CSCs exhibit a higher resistance to standard chemotherapy [39] and radiation therapy [40] through deregulated apoptosis and survival signaling. These drug-resistance properties of CSCs suggest that the majority of standard therapeutic approaches can eliminate the bulk tumor cells but may ultimately fail to obtain reliable clinical responses because conventional treatments are not as effective at eliminating CSCs; thus, the remaining CSCs are able to re-initiate tumor development in patients.
3.2. CSCs as a Novel Therapeutic Target
Despite some promising therapeutic outcomes, conventional therapeutic approaches against tumors have many limitations that frequently lead to local recurrence with subsequent metastasis and poor survival. The main reason for these cancer relapse and unsatisfactory long-term clinical responses is resistance to conventional therapy. CSC-mediated multiple drug resistance has been observed over the past half-century in various tumor types, including leukemia [41], colorectal [42], brain [43], pancreatic [44], melanoma [45], breast [46], and cancers. Moreover, CSC-mediated radioresistance was also observed in breast [47] and brain [48] cancers. Over the past years, many efforts have been devoted to investigate the potential origin of hepatic CSCs. For instance, Tang et al. found that hepatic progenitor cells can be transformed into tumor-initiating cells by transforming growth factor beta (TGFβ) and interleukin-6 (IL-6)-related signaling pathways [49]. Consistently, Wu et al. also revealed that a small subset of hepatic progenitor cells express tumor initiating cell markers during hepatocarcinogenesis in both rat and human models, and they are transformed through miR216a and Akt-dependent pathway [50]. Another study also investigated pathological characteristics of hepatic oval cells (HOCs) and their potential roles during the progression of HCC [51]. Dumble et al. showed that HOCs were involved carcinogenesis of HCC through p53 signaling pathway [52]. Likewise, c-myc expression may promote the hepatocarcinogenesis of HOCs [53]. In addition, the infection of hepatitis B or C virus (HBV or HCV) significantly increases the malignant transformation into HCC by approximately 15-to 20-fold compared with HCV-negative subjects [54]. HBV infection facilitates the expressions of various CSCs-associated transcription factors (c-Myc, Klf4, Nanog, Oct4, and Sox2) and CSCs-related genes (CD90, CD117, and CD133), and thus stimulates the self-renewal capacity of hepatocyte derived cells [55]. Similarly, Wang et al. also found that the overexpression of hepatitis B virus X protein enhances the stem-like properties and tumorigenic potential of OV positive liver CSCs by activating the MDM2/CXCR4/OV6 signaling cascades [56]. In this context, the development of novel therapeutic strategies that selectively eliminate CSCs and leave the normal and healthy cells largely unaffected is urgently required. An improvement can potentially be achieved by the selective targeting of subtle differences in surface antigens regulating their functions as well as alterations in signaling pathways of CSCs. Since their identification in multiple solid tumors and leukemia, various CSC elimination strategies selectively targeting CSC-specific surface markers and signaling pathways have been applied. While most are still at the preclinical stage, currently, some of these strategies can successfully eliminate CSCs and thereby prevent local recurrence with subsequent metastasis. The potential origin of hepatic CSCs is summarized in Figure 1.
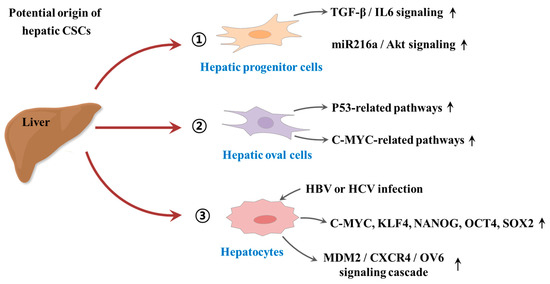
Figure 1. Schematic diagram summarizing the potential origin of hepatic cancer stem cells (CSCs). Hepatic progenitor cells can be transformed into tumor-initiating cells by activation of transforming growth factor beta (TGFβ)/interleukin-6 (IL-6)-related signaling and miR216a/Akt-dependent signaling pathways. Hepatic oval cells (HOCs) were involved carcinogenesis of hepatocellular carcinoma (HCC) through p53 and c-myc related signaling pathways. In addition, the infection of hepatitis B or C virus (HBV or HCV) significantly increases the malignant transformation into HCC by enhancing the expressions of CSCs-associated transcription factors or activating MDM2/CXCR4/OV6 signaling cascades. “↑” means increase.
3.3. The Hepatic CSC Microenvironment
Hepatic cancer occurs more frequently in patients with chronic liver diseases due to the chronic inflammatory response and continuous hepatocyte destruction/regeneration that occurs [57]. A variety of physiological changes that take place during long-term liver regeneration and inflammation can enhance both the initiation and promotion phases of hepatocarcinogenesis. These changes accelerate the accumulation of genome instability through genetic and epigenetic alterations, expansion of resident hepatic stem/progenitor cell populations, and modification of the hepatic microenvironment. Chronic liver diseases can also induce proliferation of hepatic stem/progenitor cells [58]. Their recruitment, proliferation, and development are tightly regulated by various factors transmitted from stem cell niches referred to as a specialized microenvironment [59]. The liver microenvironment is drastically changed in chronic liver diseases to favor tumors, including increased expansion of hepatic progenitor cells and endothelial progenitor cells, hepatic infiltration by lymphocytes, and the activation of hepatic stellate cells. During chronic liver diseases, hepatic stellate cells are activated and proliferate, which results in scar formation and fibrosis with excessive extracellular matrix (ECM) deposition [60]. These dynamic physiological conditions can cooperatively affect liver tumorigenesis by supporting hepatic CSC development. For instance, compared with normal fibroblasts, cancer-associated fibroblasts (CAFs) have enhanced self-renewal capacity and increased secretion of various growth factors, such as CXCL12, hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF), and vascular endothelial growth factor (VEGF), which can promote tumorigenesis [61]. Multiple growth factors or cytokines secreted by endothelial cells (ECs) and CSCs in the tumor microenvironment can promote the transformation of normal fibroblasts into CAFs [62]. Subsequently, transformed CAFs can stimulate the stem-like properties of hepatic CSCs by modulating autophagy [63]. Similarly, myofibroblast activation releases several growth factors and cytokines that may result in sustained tumor progression [64]. Mesenchymal stem cells (MSCs) have been implicated in promoting cancer cell growth, invasion/metastasis, vasculogenesis, and immunosuppression within tumor microenvironment for the restoration of cancer stem cells [65,66] by secreting various growth factors, cytokines, chemokines, and ECM components [67]. Indeed, Mi et al. found that considerable amount of IL-6 was secreted by MSCs and subsequently promoted human HCC invasion by activating IL-6/STAT3 signaling pathway [68]. In addition, the tumor microenvironment is characterized by chronic inflammatory conditions, which can promote tumor cell growth, survival, invasion, and metastasis [69]. Lymphocytic infiltration can cause the release of inflammatory molecules and the formation of oxygen free radicals, which results in DNA damage and other stresses that can stimulate tumor growth [70].
3.4. The Effect of Chemotherapy/Radiotherapy on Hepatic CSCs
CSCs have been known to exhibit various genetic and/or epigenetic alternations that are associated with the resistance to classical therapeutic strategies, such as chemotherapy and radiotherapy [71]. These various alternations include dysregulation of ATP-binding cassette (ABC) membrane transporters, cell cycle arrest (quiescent state), enhanced DNA repair efficiency, and high resistance to anticancer drug-induced apoptosis [72]. Radiation and many types of chemotherapeutic agents exert their anticancer effects by inducing DNA damage to cancer cells; thus, it seems reasonable to hypothesize that the resistance of CSCs to classical therapeutic approaches may be due to the increased expression of DNA repair-related genes, such as BRCA1 and RAD51 [73]. One of the most potent regulators of CSC resistance to DNA damaging chemotherapeutic drugs is DNA damage checkpoint protein kinases (CHKs), which are activated by genotoxic stress and delay the cell cycle progression to facilitate DNA repair [74]. Lee et al. found that depletion of 14-3-3ζ, which regulates cell cycle, differentiation, and apoptosis, increases the sensitivity to radiation therapy in CD133+ Huh7 liver cancer stem cells [75]. Ma et al. reported that CD133+ hepatic CSCs exhibit greater chemoresistance than CD133− subpopulation by activating well-known pro-survival Akt/PKB and anti-apoptotic Bcl-2 signaling pathways [76]. Another important regulator of the DNA repair systems against both endogenous and exogenous sources of DNA damage in stem cells is ATP-binding cassette transporters (ABC transporters), which can selectively extrude various toxic substrates, leading to multidrug resistance (MDR) [77]. Indeed, Fung et al. found that enhanced expression levels of ABC transporters significantly promote chemoresistance, epithelial–mesenchymal transition (EMT) and cancer stemness in HCC model [78]. PI3K/Akt, which is one of the most potent prosurvival signaling pathways, contributes to the maintenance and survival and also triggers endogenous drug resistance in CSCs [79]. Indeed, Kahraman et al. showed PI3K/Akt/mTOR pathway-mediated resistance to Rapamycin to Sorafenib cotreatment in CD133+/EpCAM+ hepetic CSCs [80]. Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) plays an important role in cancer therapy by inducing selective apoptosis of cancer cells while having little effect on the normal cells [81]. Zhu et al. reveal that TRAIL mediates drug resistance in various hepatic CSC models (PLC, HepG2 and Huh7 LC cells) through PI3K/Akt/Bad signaling cascades [82]. Another promising target molecular to induce apoptosis in CSCs is nuclear factor kappa B (NFκB), which is known as an antiapoptotic signal transcription factor, can be activated by various chemodrugs including sorafenib [83]. Zou et al. showed that sorafenib-induced NF-κB activation contributes to the enhanced resistance to sorafenib in CD133-positive sphere-forming hepatic CSCs [84]. The multidrug resistance mechanisms of hepatic CSCs are summarized in Figure 2.
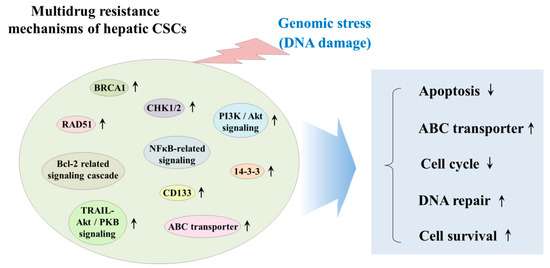
Figure 2. Schematic diagram summarizing the multidrug resistance mechanisms of hepatic CSCs. CSCs have been known to exhibit various genetic and/or epigenetic alternations, which are related to the resistance to classical therapeutic strategies, such as chemotherapy and radiotherapy. These various alternations include dysregulation of ATP-binding cassette (ABC) membrane transporters, cell cycle arrest (quiescent state), enhanced DNA repair efficiency, and high resistance to anticancer drug-induced apoptosis. “↑” means increase; “↓” means decrease.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12102746
This entry is offline, you can click here to edit this entry!