1. Introduction
1.1. Diabetes and Skeletal Muscle Insulin Resistance
Diabetes mellitus has a complex pathophysiology that combines impaired metabolism and deficient glucose disposal; it affects multiple organs and increases the risk of life-threatening cardiomyopathy, as well as complications of nephropathy, neuropathy, and retinopathy [
1,
2,
3,
4]. The skeletal muscle is the largest organ in the body and is essential to maintain vital functions such as movement, postural support, breathing, and thermogenesis [
5]. Notably, skeletal muscle is also a primary site for glucose uptake; indeed, euglycemic hyperinsulinemic clamp experiments demonstrate that 80–90% of infused glucose is taken up by skeletal muscle [
6]. Diabetes mellitus is broadly divided into type 1 (T1D) and type 2 (T2D) diabetes. T1D is a chronic autoimmune disorder in which dysfunctional pancreatic islet β-cells are targeted for destruction, thereby depleting insulin and impairing glucose uptake by peripheral tissues such as skeletal muscle and fat. This dysfunction results in persistent high circulatory glucose levels. In T2D, which accounts for about 90% of all diabetes cases, peripheral organs, including skeletal muscle, fat, and the liver, become insulin resistant, thereby leading to poor glucose clearance and high circulatory glucose levels. As skeletal muscle is the predominant site of postprandial glucose clearance, skeletal muscle insulin resistance is thought to be the major underlying cause of T2D. The persistently higher levels of circulating glucose in T2D signal pancreatic islet β-cells to produce more insulin, and eventually, the overworked β-cells become dysfunctional and insulin secretion is impaired. Thus, the skeletal muscle and pancreatic β-cells are central regulators of glucose homeostasis in the body.
Insulin resistance, also known as prediabetes, is an intermediate metabolic state between normoglycemia and T2D, wherein impaired fasting glucose and/or impaired glucose tolerance leads to metabolic dyshomeostasis. Within approximately five years of diagnosis, prediabetic individuals have an about 50% chance of developing T2D and other metabolic complications that ultimately decrease their lifespan [
7].
1.2. Skeletal Muscle Myokine-Mediated Regulatory Actions
Skeletal muscle secretes numerous myokines, which are defined as cytokines and peptides that are produced and released by muscle fibers. Myokines are involved in the autocrine regulation of metabolism in muscles and the para/endocrine regulation of other organs that express myokine receptors, including the pancreas, adipose tissue, liver, heart, bone, gut, and brain [
15,
16,
17]. For instance, myokines produced by muscles during contraction can improve insulin sensitivity and glucose oxidation via autocrine action [
18]. Furthermore, muscle fiber-derived myokines are involved in the autocrine/paracrine regulation of satellite cells and promote muscle hypertrophy during exercise [
19,
20]. Myokines involved in metabolic regulation can also ameliorate multiple diseases including insulin resistance, obesity, and cancer [
21,
22,
23,
24,
25]. Over 3000 possible myokines have been identified in humans and rodents [
26]. Interestingly, the functions of more than 100 myokines, including many novel ones, from the secretomes of primary human myotubes [
27,
28,
29] and murine myocytes [
28,
30,
31] have been determined.
Dysfunctional myokine secretion plays a role in the pathogenesis of aging and metabolic diseases, including obesity, T2D, and sarcopenia [
32,
33,
34]. Aging is associated with decreases in the secretion of beneficial myokines in rodents and humans, such as Apelin, Decorin, β-Aminoisobutyric acid (BAIBA), Sesterin, Secreted protein acidic and rich in cysteine (SPARC), Interleukin-15 (IL-15), and Irisin [
35,
36,
37,
38,
39,
40]. Furthermore, increased levels of the detrimental myokine, myostatin, is found at higher levels in streptozotocin-induced T1D mice and in the serum of patients with T1D and T2D [
41,
42,
43]. Moreover, myostatin inhibition by adeno-associated virus-induced overexpression of the myostatin propeptide in mice increased the skeletal muscle glucose uptake in insulin-resistant HFD-fed mice. Myostatin also suppresses muscle regeneration, and this pathological effect is partially reversed by regular exercise and physical activity [
44].
Myokines may be critical regulators of age-related pathologies including diabetes, muscular atrophy, and chronic inflammation. Indeed, serum from T2D patients contains reduced levels of beneficial myokines such as Irisin, IL-13, and FSTL-1 [
45]. Interestingly, myokines secreted by myotubes impact β-cell function, proliferation, and survival; myokines from healthy myotubes act in a beneficial way, while myokines from insulin-resistant myotubes act in a detrimental way, suggesting that skeletal muscle-to-pancreas cross-talk regulates insulin secretion [
46]. Similarly, the myokine expression pattern in the secretome of T2D patients differs from that of healthy individuals, and proteomic analysis from human primary skeletal muscle cells isolated from T2D patients shows altered myokine profiles compared to skeletal muscle cells from healthy donors [
47].
2. Myokine-Mediated Muscle-to-Muscle and Muscle-to-Pancreas Communication
The evidence described in the prior section indicates that skeletal muscle can communicate with other organs through myokines secreted into the bloodstream during muscle contraction. Moreover, some of the beneficial circulating myokines involved in metabolic regulation are downregulated in T2D individuals. Hence, it is no surprise that common risk factors such as a sedentary lifestyle and obesity are correlated with decreased muscle contraction, impaired energy metabolism, and insulin resistance. Therefore, strategies to improve/regulate myokine release and function could present therapeutic opportunities to prevent and/or reverse T2D.
Myokines Mediate Muscle-to-Muscle Cross Talk
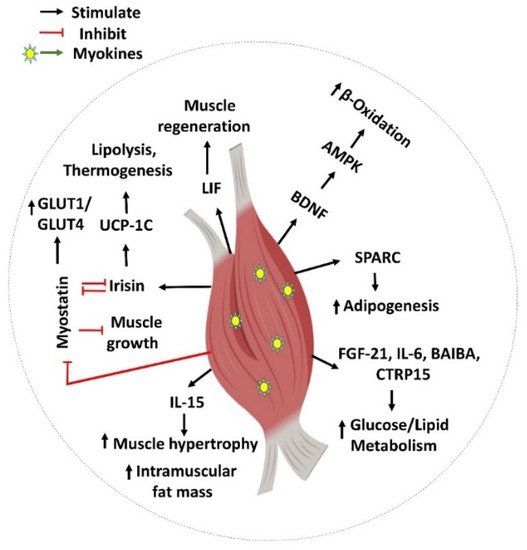
Exercise is a proven lifestyle intervention for the treatment of T2D. Improved insulin sensitivity and glucose disposal is the well-known underlying molecular mechanism for the benefits of physical activity on T2D. Myokines released during or after exercise, which can exert effects locally within the muscle, are emerging as key mechanisms for these muscle metabolic modifications (Figure 1). Most of these secreted myokines influence metabolism, and/or are involved in muscle regeneration, satellite cell proliferation, and hypertrophic responses. Therefore, myokines are important for regulating skeletal muscle homeostasis and its adaptation to exercise training.
Figure 1. Myokine-mediated regulation of skeletal muscle function. Myokines such as SPARC, FGF-21, IL-6, BAIBA, CTRP15, BDNF, LIF, Irisin, myostatin (GDF-8), and IL-15 are involved in various biological processes including muscle generation, adipogenesis, muscle hypertrophy, muscle growth, and glucose and lipid regulation locally inside the skeletal muscle. This figure was created with Biorender.com.
FGF21: Fibroblast growth factor 21 (FGF21) is a myokine with multiple therapeutic benefits against obesity-related medical complications [
48]. The activity of FGFs is mediated by their binding to FGF receptors (FGFRs) and the co-receptor β-Klotho (KLB) [
49,
50]. In vivo gene knockout and activating antibodies for FGFR1 or the FGFR1/KLB complex determined that the FGFR1C isoform is an important target of FGF21′s function [
51,
52,
53]. FGF21 expression in human skeletal muscle is reported to be activated during hyperinsulinemia, and thus it has been classified as a novel insulin-stimulated myokine [
54]. FGF21 mRNA and protein levels were reported to be increased in the gastrocnemius muscle and serum of skeletal muscle-specific AKT1-overexpressing mice. In addition, AKT-enriched C2C12 myotubes showed elevated FGF21 expression [
55]. Both of these results indicate that FGF21 secretion by skeletal muscle is regulated by the phosphatidylinositol 3-kinase (PI3-kinase)/AKT1 signaling pathway.
FGF21 regulates glucose and lipid metabolism and helps in maintaining energy balance. In support of this notion, FGF21 injection lowers fasting glucose, triglycerides, insulin, and glucagon levels in obese diabetic rodents [
56,
57] and rhesus monkeys [
58,
59]. Furthermore, chronic administration of FGF21 analogs ameliorates dyslipidemia and reduces body weight in obese and T2D patients, and also decreases fasting insulin levels while increasing adiponectin levels [
60,
61]. Acting via AMPK regulation, FGF21 protects against atrophy-induced inflammation, and its deficiency induces inflammation and worsens the obesity-induced atrophy of skeletal muscle [
62]. Thus, overall, FGF21 is an insulin-stimulated beneficial myokine that regulates energy metabolism and protects against chronic metabolic disorders such as T2D and obesity.
Irisin: Irisin is a beneficial myokine secreted by contracting skeletal muscle into the circulation after proteolytic cleavage from its precursor, fibronectin type III domain-containing protein 5 (FNDC5) [
63]. Mice overexpressing FNDC5 exhibited protection from high fat diet (HFD) diet-induced insulin resistance [
64]. FNDC5 is regulated by a peroxisome proliferator-activated receptor γ coactivator 1-α (PGC1α) [
65], a master regulator of genes involved in metabolism, thermogenesis, and antioxidant defense. In response to exercise, PGC1α expression and activity levels are elevated, and it coordinates the regulation of nuclear- and mitochondrial-encoded genes needed for contractile and metabolic adaptations in skeletal muscle [
66,
67,
68]. Consistent with this, FNDC5 protein expression was increased in muscle obtained from exercise-trained rodents and humans, whilst plasma Irisin levels were shown to be increased in mice and humans after endurance exercise [
64]. In addition, using adenoviral overexpression of FNDC5, the same study had reported that Irisin increases total body energy expenditure and protects against obesity-induced insulin resistance in mice.
Moreover, recent clinical studies have shown that circulating Irisin levels are reduced in T2D patients [
69,
70]. Consistent with this, ex vivo Irisin treatment improved the insulin-stimulated glucose uptake in muscle cells exposed to a lipotoxic T2D-mimicking milieu containing high palmitate levels [
71]. Irisin’s effects are mediated by AMPK activation, which triggers p38 MAPK signaling and GLUT4 vesicle trafficking to the plasma membrane [
72,
73]. Despite many reported beneficial effects, the receptor for Irisin still remains unknown in most of the tissues except osteocytes, adipocytes, and enterocytes where αVβ5 integrin is determined as the Irisin receptor [
74]. Overall, it has been reported that Irisin regulates glucose metabolism in skeletal muscle in an autocrine manner [
73]. Given that Irisin also has positive effects in physiological functions such as thermogenesis, and glucose- and lipid-oxidation, it carries potential to be an attractive target for treating metabolic disorders.
SPARC: Secreted protein acidic and rich in cysteine (SPARC)/osteonectin is an exercise-responsive myokine. It has been reported that exercise-induced changes in muscle performance (metabolic strength and development), including lactate-induced changes, are SPARC-dependent [
75]. For example, whole-body SPARC knockout mice exhibited an impaired metabolism and defective phosphorylation of AMPK and protein kinase B in the skeletal muscle. Consistent with this, treatment with SPARC (injected intraperitoneally with recombinant SPARC protein) improved glucose tolerance and activated AMPK in the skeletal muscle of SPARC knockout mice [
76]. In addition, SPARC treatment to HFD-induced obese mice reversed their glucose intolerance and restored skeletal muscle AMPK signaling. SPARC deficiency in mice also decreases skeletal muscle mass and increases age-dependent adiposity, as skeletal muscle mass changes are inversely correlated with adipose mass changes [
77]. In cultured myoblasts, SPARC treatment induces myogenic differentiation [
78,
79]. SPARC gene expression is reduced during aging, which may be related to observed age-related decreases in the levels of skeletal muscle progenitor cells [
80]. Overall, SPARC is a beneficial myokine that is involved in AMPK-mediated glucose regulation and improves glucose tolerance.
BAIBA: Known also as 3-amino-2-methylpropanoic acid, BAIBA is a small molecule catabolite of thymine and valine metabolism in mammals, which is produced by and secreted from skeletal muscle. BAIBA is a novel protective myokine that is increased during exercise via a PGC1α-dependent mechanism, improves insulin sensitivity, and protects against HFD-induced obesity [
81,
82]. Similar to other myokines, BAIBA enrichment/overexpression increases fatty acid oxidation and decreases lipogenesis in mice, resulting in a reduced body fat percentage [
83]. BAIBA is produced in skeletal muscle during exercise and protects against obesity-dependent metabolic disorders, including T2D and non-alcoholic fatty liver disease [
84,
85]. BAIBA treatment of palmitate-exposed C2C12 myocytes and the skeletal muscle of HFD-fed mice ameliorated defects in the insulin receptor substrate (IRS)-1/Akt-mediated insulin signaling pathway. In addition, BAIBA infusion reversed HFD-induced weight gain and improved glucose tolerance in mice. BAIBA also suppressed inhibitory κBα (IκBα) phosphorylation, nuclear factor κB (NFκB) nuclear translocation, whilst promoting AMPK phosphorylation and the expression of peroxisome proliferator-activated receptor gamma (PPARδ) in mouse skeletal muscle and C2C12 cells [
82]. Thus, BAIBA treatment protects against insulin resistance, prevents inflammation, and improves β-oxidation in skeletal muscle via the AMPK-PPARδ pathway. As with most other myokines discussed so far, BAIBA also communicates in a paracrine fashion, whereby it enhances the browning of white adipose tissue and increases β-oxidation in the liver through mechanisms mediated by peroxisome proliferator-activated receptor α (PPARα) [
83]. Thus, BAIBA treatment prevents HFD-induced obesity through improving glucose tolerance, β-oxidation, and suppressing inflammatory pathways [
81,
86].
Brain-derived neurotrophic factor (BDNF): Protein and mRNA levels of BDNF are increased in human skeletal muscle after exercise [
87]. BDNF is abundantly expressed in slow twitch skeletal muscle fibers, and its beneficial effects in skeletal muscle are mediated through AMPKα-PGC1α-mediated mitochondrial function and β-oxidation [
88]. BDNF initiates its beneficial effects by binding to the tropomyosin-related kinase receptor B (TrkB), which subsequently activates phosphoinositide-3-kinase (PI3K)/Akt, Ras/extracellular signal-regulated kinase (ERK), and phospholipase C (PLCγ)/protein kinase C (PKC) signaling pathways [
89]. Skeletal muscle specific BDNF knockout mice have impaired glucose to fatty acid utilization during fasting, linked to reduced muscle strength, myofiber necrosis and insulin resistance [
90]. Interestingly, skeletal muscle-specific BDNF regulates the glycolytic muscle fiber type and metabolism [
91]. BDNF addition to C2C12 myotubes correlates with a high mitochondrial DNA content and increased β-oxidation rate, facilitating mitochondrial fatty acid transport. Similarly, chronic subcutaneous or intracerebroventricular administration of BDNF increased muscle glucose uptake and enhanced energy expenditure in obese diabetic C57BL/KsJ-db/db mice [
92]. Together, these pieces of evidence indicate that BDNF signaling is vital for balancing glucose and lipid metabolism in skeletal muscle.
Interleukin-6 (IL-6): IL-6 is synthesized by and released from skeletal muscle in large amounts during physical activity, classifying it as a myokine. However, disparate reports of IL-6 contributing to positive and negative actions have led to controversy. For example, one finding that IL-6 pre-treatment in mice improves skeletal muscle glucose uptake, as assessed by hyperinsulinemic-euglycemic clamp analysis [
93], supports the concept that IL-6 plays a positive role in skeletal muscle. In addition, at 3 months of age, IL-6 knockout mice showed an impaired exercise capacity and glucose intolerance, and they became obese by 9 months; however, these anomalies were linked to decreased levels of AMPK, making it unclear whether IL-6 was the causative factor [
94]. Consistent with a beneficial effect of IL-6, in humans, IL-6 injection stimulated GLUT4 translocation and improved skeletal muscle insulin sensitivity [
95]. Counterintuitively, IL-6 levels can be found elevated in insulin resistance and T2D. In addition, palmitate-induced IL-6 production was associated with a decreased glucose uptake in myocytes; this was reversed by an anti–IL-6 antibody [
96,
97]. Further confounding the interpretation of IL-6 function, IL-6 production is stimulated by TNFα and was initially found to be elevated in T2D [
98], yet a recent human study found no changes in the circulating levels of IL-6 in T2D patients compared to control subjects [
99]. Overall, IL-6 is stimulated by physical activity, but its effect on T2D is less clear, with evidence of both positive and negative actions.
Leukemia inhibitory factor (LIF): LIF is produced by and released from skeletal muscle cells [
100]. Recombinant human LIF induces myoblast proliferation, and LIF mRNA and protein levels were found to be upregulated in contracting cultured human myotubes isolated from muscle biopsies of the vastus lateralis muscle, as well as in human skeletal muscle after resistance exercise [
101]. LIF activates the transcription factors Jun-B and c-Myc, which promote satellite cell proliferation in an autocrine or paracrine fashion [
101]. LIF was also found to increase the phosphorylation of AKT at Ser
473 in soleus and extensor digitorum longus muscles and increase glucose uptake in both oxidative and glycolytic muscles [
102]. Counterintuitively, LIF protein and its receptor (LIFR) are also elevated in muscle tissue and cultured myoblasts from T2D individuals, but LIF-stimulated cell proliferation is impaired in diabetic myoblasts [
103,
104]. Given that others have reported that LIF is immediately secreted and does not accumulate in skeletal muscle [
105], it remains possible that these disparate findings could be caused by secretion defects in diabetic individuals rather than increased LIF biosynthesis. Experiments that distinguish these possibilities will be important to gain a deeper understanding of the interplay between LIF and metabolic disease.
Interleukin-15 (IL-15): Skeletal muscle is an important source of circulatory IL-15 levels. IL-15 is a member of the IL-2 superfamily and, in humans and mouse models, IL-15 levels increase after acute physical exercise [
35,
106,
107,
108]. IL-15 is associated with beneficial actions; for instance, IL-15 overexpression induces weight loss and reduces white adipose tissue mass in rodents [
95,
109,
110]. Moreover, enrichment of IL-15 protects against HFD-induced obesity and insulin resistance in mice models [
111,
112]. Consistent with this, obese human subjects have decreased levels of circulating IL-15 compared to lean individuals [
95]. However, although IL-15 treatment of C2C12 myotubes increases GLUT4 gene expression and GLUT4 vesicle translocation, glucose uptake is not coordinately increased [
113,
114]. Instead, the effect of IL-15 is likely to occur at the level of the muscle tissue. In rodents, increased levels of circulating IL-5 induced fiber-type shifts, which promote an oxidative phenotype with increased mitochondrial DNA levels and cytochrome C oxidase activity [
35,
115]. Indeed, IL-15 therapy was found to mimic the anti-aging effects of exercise on skeletal muscle and skin in mouse models, suggesting it is a beneficial strategy to attenuate aging [
35]. Furthermore, IL-15 treatment of skeletal muscle cells was found to exert protection against H
2O
2-induced oxidative stress and enhance mitochondrial function through a PPARδ-dependent mechanism. Overall, IL-15 may act in an auto/paracrine manner that is responsible for the skeletal muscle-mediated positive effects of exercise. Collectively, this evidence suggests that increasing IL-15 expression is a candidate intervention to prevent and remediate obesity and T2D.
Myonectin (CTRP15): Myonectin is a recently discovered mycophenolate that is released by skeletal muscle. It belongs to the C1q/TNF-related protein (CTRP) family, which is involved in the regulation of glucose and fatty acid metabolism [
116,
117,
118]. Amongst the CTRP family members, myonectin is the one whose expression is limited only to skeletal muscle [
119]. Moreover, slow-twitch fibers with higher oxidative metabolism express higher levels of the myonectin gene relative to fast-twitch fibers, which have a higher glycolytic metabolism. Elevated levels of intracellular calcium have been shown to increase the expression of myonectin in skeletal muscle [
120,
121]. Myonectin is elevated in adults with T2D and increased adiposity, relative to healthy individuals, likely as a compensatory mechanism against insulin resistance [
122]. However, diet-induced obesity in mice does not cause this compensatory mechanism—the muscle mRNA levels and circulating protein levels of myonectin were reduced relative to control mice, and subsequent voluntary exercise increased myonectin gene expression and circulating protein levels [
123]. This conundrum was resolved when it was determined that myonectin levels are raised after feeding, indicating that myonectin secretion could be regulated by substrate availability. For example, overnight fasting decreases myonectin levels, and subsequent feeding with glucose or emulsified lipids increases circulating myonectin levels in mouse models [
123]. Overall, myonectin is an important mediator in inter-organ cross-talk and its secretion by skeletal muscle increases with the higher availability of glucose and fatty acids in the insulin-resistant and T2D state as a compensatory mechanism to improve glucose tolerance and increase fatty acid oxidation [
122,
124].
Myostatin: Myostatin, also named growth and differentiation factor-8 (GDF-8), is expressed in both embryonic and adult skeletal muscle. It is secreted by skeletal muscle and cardiac cells and is reported to inhibit muscle growth and differentiation and reduce skeletal muscle mass [
125,
126]. Consistent with this, myostatin-suppressed mice and cattle are larger than control animals, suggesting that myostatin functions as a ‘brake’ to suppress skeletal muscle growth [
127,
128]; similar findings have been reported for humans and dogs [
129]. Myostatin is a member of the transforming growth factor β (TGFβ) superfamily. Mechanistically, myostatin binds to activin type IIA and IIB receptors (ActRIIA/B) and TGFβ receptors (TGFβRII) at the plasma membrane. The myostatin-mediated muscle growth impairment is caused by activating activin, which in turn phosphorylates SMAD2/3 and promotes the establishment of a heterotrimeric complex with SMAD4 [
130]. Furthermore, the inhibition of myostatin-induced reactive oxygen species (ROS) is an effective treatment for reducing muscle wasting during sarcopenia [
131]. Interestingly, myostatin ablation in mice skeletal muscle was also discovered to prevent fat mass gain [
132]. While myostatin was initially discovered as a myokine, it was later determined to also be secreted by adipose tissue, and thus is termed as an adipo-myokine [
133]. Consistent with a role in adipose tissue, myostatin knockout mice had shown a reduced fat pad mass and were resistant to obesity and insulin resistance [
134,
135,
136]. Further, the inhibition of myostatin, via a loss-of-function mutation in one or both alleles of the
myostatin gene, improves whole-body insulin sensitivity and alleviates the development of insulin resistance in obese mice; the genetic loss of myostatin also improves insulin sensitivity and glucose tolerance in severely obese mouse models [
135,
137,
138]. Moreover, the muscle-specific inhibition of myostatin increases the protein levels of GLUT1 and GLUT4 in rat muscle [
139], providing a mechanistic basis for the beneficial effects of myostatin inhibition to improve glucose tolerance. Together, myostatin is a negative myokine/adipokine that impairs glucose uptake, enhances adiposity, and impairs muscle growth and function.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23094636