Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Serine and cysteine proteases are a class of drug targets that could greatly benefit from the features of the nitrile group as a warhead for the design and discovery of innovative and effective drugs.
- nitrile
- warhead
- cysteine/serine/threonine protease
1. Inhibitors of Viral Cysteine Proteases
Enterovirus 71 (EV71) belongs to the Picornavirus family of viruses. It causes neurological diseases in adults and the hand, foot, and mouth disease in children. Outbreaks of EV71 have emerged as a global health issue due to severe neurological complications leading to fatal encephalitis in a relevant number of cases. The viral genome, constituted by a single-stranded RNA molecule, is translated into a large polyprotein inside the host cell. The virally encoded protease 3Cpro plays a crucial role in the proliferation of this virus and represents a promising drug target for the design of antiviral drugs [1][2].
Wang et al. investigated different warheads in order to improve the stability and selectivity of a previously developed set of inhibitors of the 3Cpro enzyme of EV71 that were originally designed with an aldehyde warhead [3][4]. They introduced a panel of warheads, such as nitrile, ketoamide, alcohol, hydroxyl amide, doubly activated olefins, and hydroxy-nitrile groups, as an alternative to the aldehyde group. In this case, the nitrile warhead led to a moderately active inhibitor (4), while an α-hydroxynitrile gave better selectivity toward cysteine vs. serine protease and cell-based activity (inhibitor 5, Figure 1). An X-ray crystal structure of the enzyme-bound inhibitor revealed a non-covalent binding mode [4].
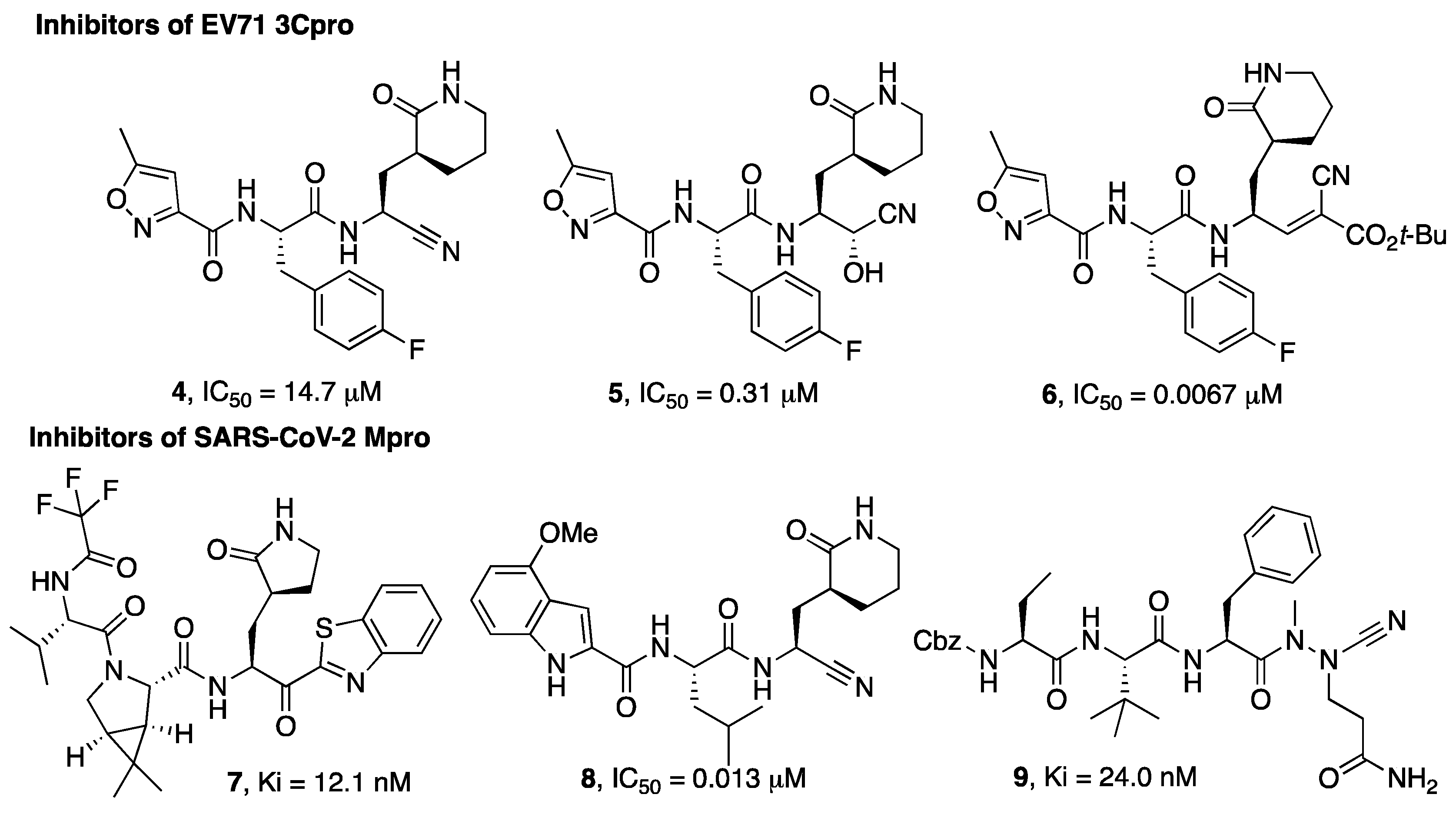
Figure 1. Chemical structures of viral cysteine proteases inhibitors 4–6 (EV71 3Cpro) and 7–9 (SARS-CoV-2 Mpro).
A different example of a warhead was introduced in compound 6. The electron-withdrawing properties of the cyano group were exploited for the preparation of double-activated olefins that are able to function as Michael acceptors upon reaction with the cysteine active site residue of EV71 3Cpro. The cyano group, which, in this case, does not form a covalent bond with the active site cysteine but contributes to increasing the reactivity of the olefin carbon, provided more selective enzyme inhibitory activity coupled to a better and broad-spectrum activity on cell-based assays (6) [5][6].
MPro is a cysteine protease with an important role in the SARS-CoV-2 replicative cycle, and since the SARS-CoV-2 epidemic outbreak in 2019, intense research efforts have been devoted by scientists in the search for MPro inhibitors [7][8][9]. These efforts culminated with the discovery of the drug Paxlovid, which is a combination of the Mpro inhibitor nirmatrelvir with the metabolic booster ritonavir. Nirmatrelvir (3) was developed based on a structure-based optimization approach aimed at optimizing the interaction with the S1 and S2 pockets and, at the same time, monitoring the solubility and selected pharmacokinetic properties of the designated hits. The selection of 3 to be advanced in preclinical optimization over the benzothiazole analogue 7 was mainly supported by improved water solubility and a lower propensity to epimerization at the α-carbon, attributable to the presence of the cyano group instead of the benzothiazole moiety [10].
The X-ray co-crystal structures of SARS-CoV-2 Mpro in complex with nirmatrelvir confirmed the formation of a reversible covalent Cys145 adduct with the nitrile substituent [10][11]. It is worth mentioning that this inhibitor has a similar efficacy toward the recently emerged SARS-CoV-2 variants [12].
Briefly, the crystal structure of Mpro in complex with nirmatrelvir (3) showed a full occupancy of the ligand within the active site of the enzyme, targeting the S1-S4 subsites, as illustrated in Figure 2. The nitrile functional group interacts at the S1′ subsite, forming a covalent adduct with the reactive Cys145 of the enzyme. In addition, H bonds with the backbone of Cys145 and His164 are evident. The pyrrolidinone moiety strongly targets the S1 subsite, establishing a series of H bonds with the backbone of Phe140 and the sidechains of Glu166 and His163. The azabicyclo moiety interacts at the S2 subsite by hydrophobic contacts, while the N-terminal moiety of the molecule can establish a series of H bonds with the backbone of Glu166. The portion bearing the trifluoro function targets residues at the S3 and S4 subsites, forming hydrophobic contacts.
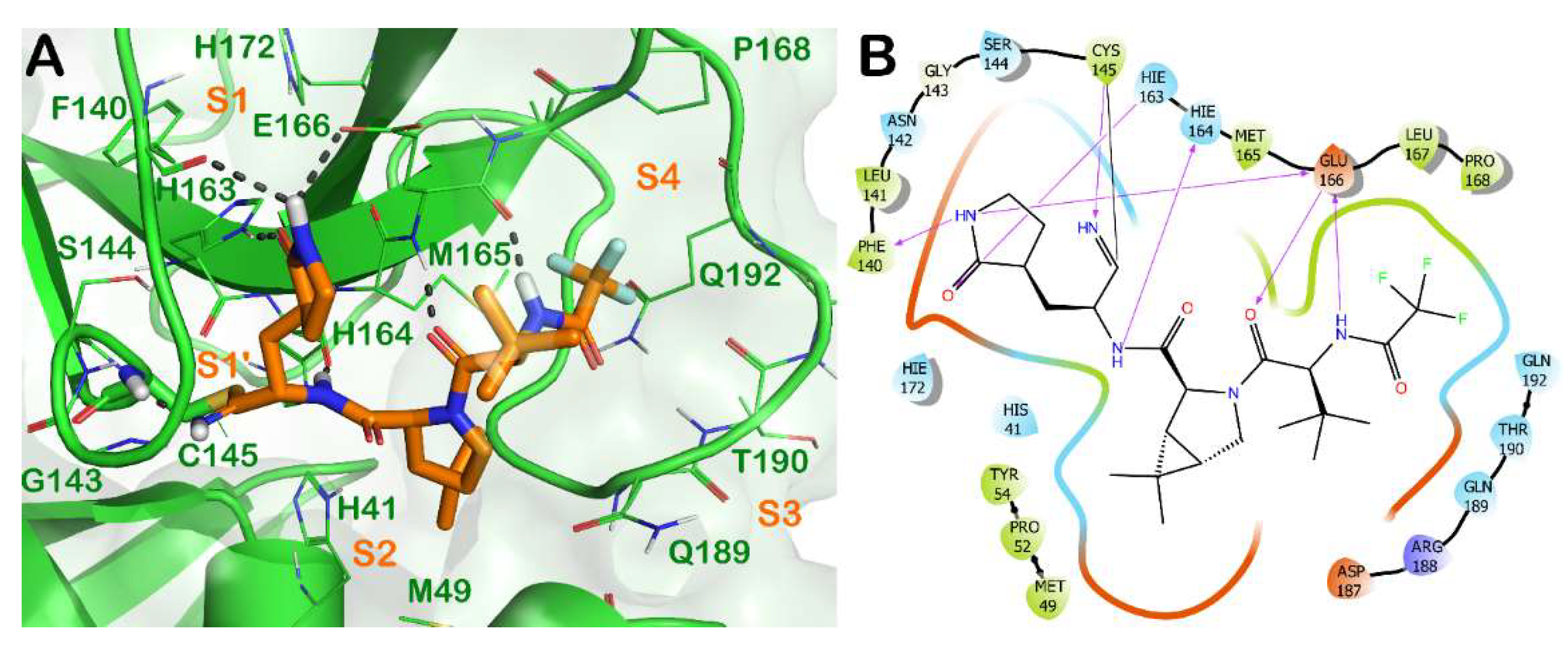
Figure 2. (A) Crystal structure of SARS-CoV-2 Mpro (green cartoon) in complex with the approved drug nirmatrelvir (3) (orange sticks) (PDB ID 7VH8). Interacting residues within the binding site are represented by lines, and the covalent bond established by Cys145 is represented by sticks. H bonds are illustrated by grey dotted lines. For the sake of clarity, water molecules are removed, and the complex was treated by the protein preparation wizard module available in Maestro (Maestro, Schrödinger LLC, release 2020-3). The picture was generated by PyMOL (The PyMOL Molecular Graphics System, v1.8; Schrödinger, LLC, New York, NY, USA, 2015). (B) Detailed interaction diagram of nirmatrelvir within the Mpro binding site, as provided by the ligand interaction diagram available in Maestro (Maestro, Schrödinger LLC, release 2020-3).
In the study by Bai et al. [13] a series of inhibitors typified by 8 (Figure 1) are described and, also in this study, the nitrile warhead was pointed out as a good alternative to the aldehyde warhead to achieve activity against the Mpro enzyme. Importantly, this study reports the selectivity of the compounds bearing a nitrile warhead vs. human cathepsins B, S, and L, suggesting that the designed compounds bearing a nitrile warhead were significantly less active in inhibiting cathepsins S and L than the hydroxymethylketon warhead, even though further studies are needed to support this observation.
A different warhead has been described by Bridenbach and colleagues [14]. In their search for potent SARS-CoV-2 Mpro inhibitors, two classes of compounds were reported, one of them being based on an azanitrile warhead (9). The authors also confirmed that these azanitrile inhibitors were able to covalently interact with the Mpro catalytic cysteine moiety.
2. Inhibitors of Protozoan Parasite Proteases
2.1. Inhibitors of Cruzain (Cz) from Trypanosoma cruzi
Cruzain (Cz) is the major cysteine protease of T. cruzi [15], the causative agent of Chagas disease, whose therapy is mainly based on the use of two drugs: benznidazole and nifurtimox. Both these drugs are responsible for serious side effects and are ineffective against the chronic stage of the disease [16]. Innovative therapeutic approaches are urgently needed for the treatment of this disease that affects one million people worldwide, with major diffusion in Latin American [17]. Cz has been explored for several years as a drug target, and nitrile-based inhibitors have been deeply investigated [15]. The most recent examples are reported below.
2.2. Inhibitors of Rhodesain (RD) from Trypanosoma brucei
Human African trypanosomiasis (HAT), also defined as sleeping sickness, is a parasitic disease that mainly affects sub-Saharan Africa. The parasite responsible for the disease is Trypanosoma brucei, for which two subspecies are known: T. b. gambiense and T. b. rhodesiense, leading to different symptomatology. The cysteine protease of T. brucei, namely rhodesain (RD), has been explored as a drug target through the design and synthesis of nitrile-based peptidomimetics [18][19].
2.3. Inhibitors of Cysteine Protease B from Leishmania spp.
Leishmaniasis is a vector-borne parasitic disease with a widespread distribution in tropical and subtropical countries, including the Mediterranean region. It has been reported that from 700,000 to 1 million new cases of leishmaniasis occur annually, while the chemotherapy for this infection is largely inadequate [20][21]. An attractive biological target for therapeutic intervention in the treatment of leishmaniasis has been identified in the cysteine protease B (CPB) expressed by this pathogenic protozoan.
2.4. Apicomplexa Parasites: Toxoplasma gondii and Plasmodium falciparum
Toxoplasma gondii is a neurotrophic protozoan parasite that exists in two principal forms, the tachyzoite and bradyzoite, this latter form being responsible for the chronic infection during which cysts form within the central nervous system (CNS) and muscle tissue to establish a life-long infection. T. gondii cathepsin protease L (TgCPL) is critical to parasite survival during the chronic phase of infection [22][23]. So, the development of effective and brain-penetrant inhibitors could lead to a great advance in the chemotherapy of this parasitic infection, which is currently ineffective against the chronic form.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27082561
References
- Diarimalala, R.O.; Hu, M.; Wei, Y.; Hu, K. Recent advances of enterovirus 71 targeting Inhibitors. Virol. J. 2020, 17, 173.
- Wen, W.; Qi, Z.; Wang, J. The Function and Mechanism of Enterovirus 71 (EV71) 3C Protease. Curr. Microbiol. 2020, 77, 1968–1975.
- Wang, Y.; Cao, L.; Zhai, Y.; Ma, J.; Nie, Q.; Li, T.; Yin, Z.; Sun, Y.; Shang, L. Inhibition of enterovirus 71 replication by an α-hydroxy-nitrile derivative NK-1.9 k. Antivir. Res. 2017, 141, 91–100.
- Zhai, Y.; Zhao, X.; Cui, Z.; Wang, M.; Wang, Y.; Li, L.; Sun, Q.; Yang, X.; Zeng, D.; Liu, Y.; et al. Cyanohydrin as an Anchoring Group for Potent and Selective Inhibitors of Enterovirus 71 3C Protease. J. Med. Chem. 2015, 58, 9414–9420.
- Ma, Y.; Li, L.; He, S.; Shang, C.; Sun, Y.; Liu, N.; Meek, T.D.; Wang, Y.; Shang, L. Application of Dually Activated Michael Acceptor to the Rational Design of Reversible Covalent Inhibitor for Enterovirus 71 3C Protease. J. Med. Chem. 2019, 62, 6146–6162.
- Liu, M.; Xu, B.; Ma, Y.; Shang, L.; Ye, S.; Wang, Y. Reversible covalent inhibitors suppress enterovirus 71 infection by targeting the 3C protease. Antivir. Res. 2021, 192, 105102.
- Ning, S.; Yu, B.; Wang, Y.; Wang, F. SARS-CoV-2: Origin, Evolution, and Targeting Inhibition. Front. Cell. Infect. Microbiol. 2021, 11, 676451.
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 M(pro): A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607.
- Banerjee, R.; Perera, L.; Tillekeratne, L.M.V. Potential SARS-CoV-2 main protease inhibitors. Drug Discov. Today 2021, 26, 804–816.
- Owen, D.R.; Allerton, C.M.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593.
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 2021, 1–5.
- Ullrich, S.; Ekanayake, K.B.; Otting, G.; Nitsche, C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg. Med. Chem. Lett. 2022, 62, 128629.
- Bai, B.; Arutyunova, E.; Khan, M.B.; Lu, J.; Joyce, M.A.; Saffran, H.A.; Shields, J.A.; Kandadai, A.S.; Belovodskiy, A.; Hena, M. Peptidomimetic nitrile warheads as SARS-CoV-2 3CL protease inhibitors. RSC Med. Chem. 2021, 12, 1722–1730.
- Breidenbach, J.; Lemke, C.; Pillaiyar, T.; Schäkel, L.; Al Hamwi, G.; Diett, M.; Gedschold, R.; Geiger, N.; Lopez, V.; Mirza, S.; et al. Targeting the Main Protease of SARS-CoV-2: From the Establishment of High Throughput Screening to the Design of Tailored Inhibitors. Angew. Chem. Int. Ed. Engl. 2021, 60, 10423–10429.
- Cazzulo, J.J.; Stoka, V.; Turk, V. Cruzipain, the major cysteine proteinase from the protozoan parasite Trypanosoma cruzi. Biol. Chem. 1997, 378, 1–10.
- Perez-Molina, J.A.; Crespillo-Andujar, C.; Bosch-Nicolau, P.; Molina, I. Trypanocidal treatment of Chagas disease. Enferm. Infecc. Microbiol. Clin. (Engl. Ed.) 2021, 39, 458–470.
- Abras, A.; Ballart, C.; Fernandez-Arevalo, A.; Pinazo, M.J.; Gascon, J.; Munoz, C.; Gallego, M. Worldwide Control and Management of Chagas Disease in a New Era of Globalization: A Close Look at Congenital Trypanosoma cruzi Infection. Clin. Microbiol. Rev. 2022, 35, e0015221.
- Dos Santos, N.I.J.; de Aquino, T.M.; da Silva-Junior, E.F. Cruzain and Rhodesain Inhibitors: Last Decade of Advances in Seeking for New Compounds Against American and African Trypanosomiases. Curr. Top Med. Chem. 2021, 21, 1871–1899.
- Kourbeli, V.; Chontzopoulou, E.; Moschovou, K.; Pavlos, D.; Mavromoustakos, T.; Papanastasiou, I.P. An Overview on Target-Based Drug Design against Kinetoplastid Protozoan Infections: Human African Trypanosomiasis, Chagas Disease and Leishmaniases. Molecules 2021, 26, 4629.
- Pradhan, S.; Schwartz, R.A.; Patil, A.; Grabbe, S.; Goldust, M. Treatment options for leishmaniasis. Clin. Exp. Dermatol. 2022, 47, 516–521.
- Caridha, D.; Vesely, B.; van Bocxlaer, K.; Arana, B.; Mowbray, C.E.; Rafati, S.; Uliana, S.; Reguera, R.; Kreishman-Deitrick, M.; Sciotti, R.; et al. Route map for the discovery and pre-clinical development of new drugs and treatments for cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2019, 11, 106–117.
- Pittman, K.J.; Aliota, M.T.; Knoll, L.J. Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection. BMC Genom. 2014, 15, 806.
- Larson, E.T.; Parussini, F.; Huynh, M.H.; Giebel, J.D.; Kelley, A.M.; Zhang, L.; Bogyo, M.; Merritt, E.A.; Carruthers, V.B. Toxoplasma gondii cathepsin L is the primary target of the invasion-inhibitory compound morpholinurea-leucyl-homophenyl-vinyl sulfone phenyl. J. Biol. Chem. 2009, 284, 26839–26850.
This entry is offline, you can click here to edit this entry!