Sphingolipids (SLs) are lipids with complex structures which were named after their sphinx-like structure by Thudichium in 1870. Lipids are major components of membranes in all eukaryotic cells determining the structural and functional integrity of cells. SLs are one of the important structural components of plasma membranes in all eukaryotes, but they also act as bioactive signaling molecules with numerous cellular physiological functions that include cell adhesion, cell proliferation, cell migration, inflammatory response and apoptosis. Sphingolipids, which act as a bioactive signaling molecules, are involved in several cellular processes such as cell survival, proliferation, migration and apoptosis. An imbalance in the levels of sphingolipids can be lethal to cells. Abnormalities in the levels of sphingolipids are associated with several human diseases including kidney diseases. Several studies demonstrate that sphingolipids play an important role in maintaining proper renal function. Sphingolipids can alter the glomerular filtration barrier by affecting the functioning of podocytes, which are key cellular components of the glomerular filtration barrier.
- sphingolipids
- glomerular diseases
- tubulointerstitial diseases
1. Sphingolipid Biosynthesis and Catabolism
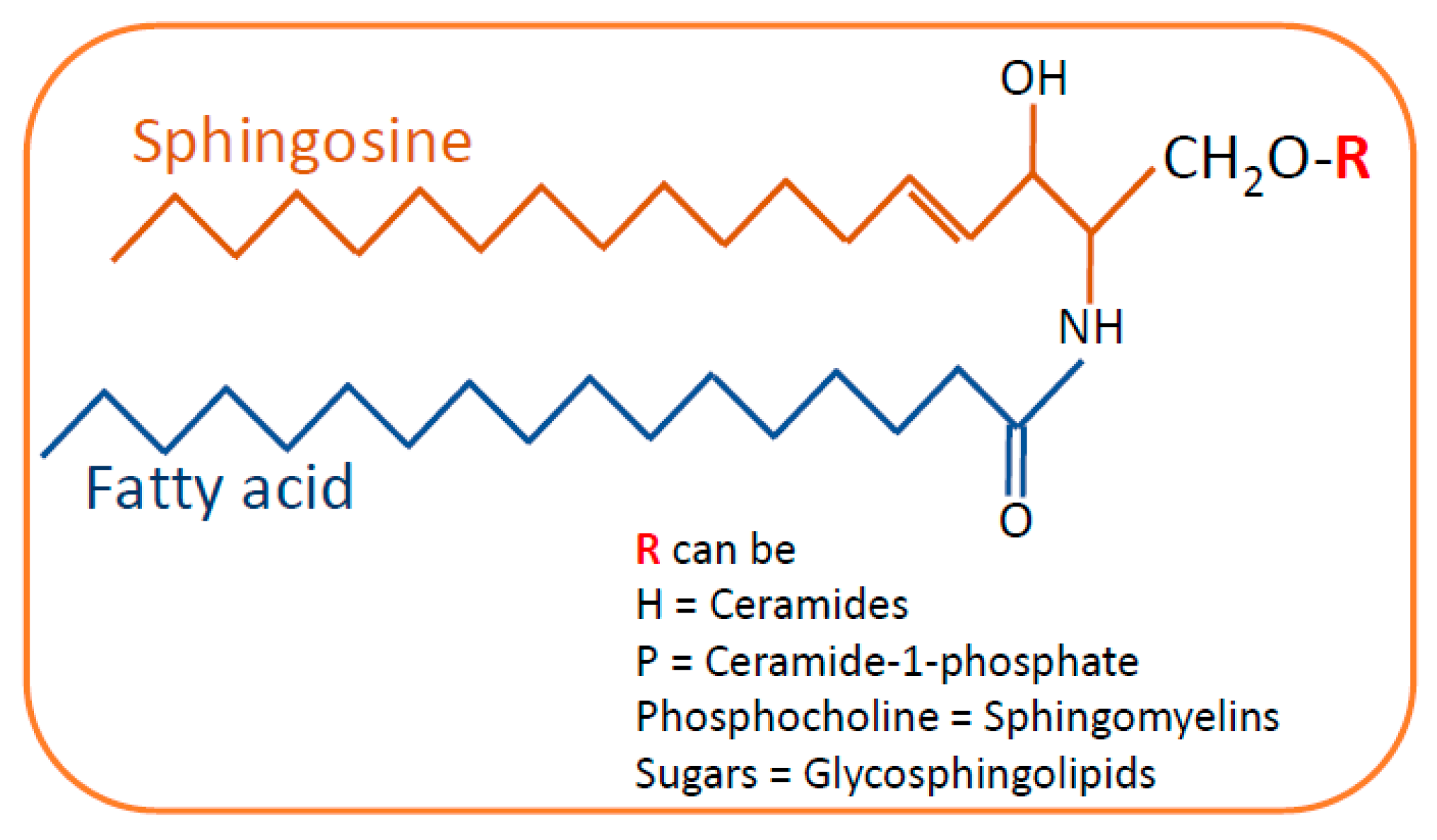
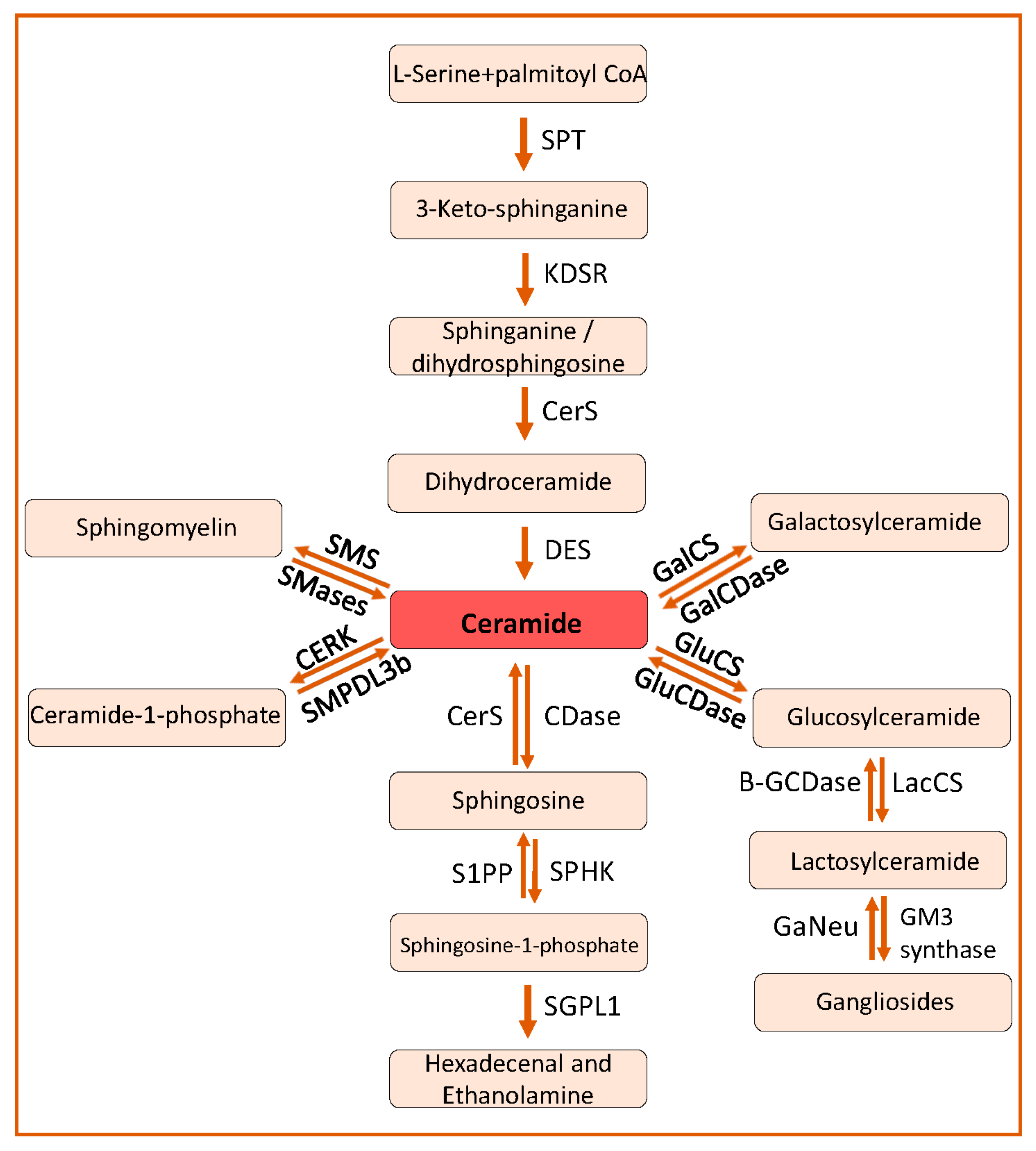
2. Sphingolipids in Kidney Diseases
2.1. Role of Sphingolipids in Podocyte Injury in Glomerular Diseases
2.2. Role of Sphingolipids in Tubulointerstitial Fibrosis and Acute Kidney Injury
This entry is adapted from the peer-reviewed paper 10.3390/ijms23084244
References
- Mallela, S.K.; Mitrofanova, A.; Merscher, S.; Fornoni, A. Regulation of the amount of ceramide-1-phosphate synthesized in differentiated human podocytes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 158517.
- D’Angelo, G.; Capasso, S.; Sticco, L.; Russo, D. Glycosphingolipids: Synthesis and functions. FEBS J. 2013, 280, 6338–6353.
- Papadopoulos, T.; Krochmal, M.; Cisek, K.; Fernandes, M.; Husi, H.; Stevens, R.; Bascands, J.-L.; Schanstra, J.P.; Klein, J. Omics databases on kidney disease: Where they can be found and how to benefit from them. Clin. Kidney J. 2016, 9, 343–352.
- Hodgin, J.B.; Nair, V.; Zhang, H.; Randolph, A.; Harris, R.C.; Nelson, R.G.; Weil, E.J.; Cavalcoli, J.D.; Patel, J.M.; Brosius, F.C.; et al. Identification of Cross-Species Shared Transcriptional Networks of Diabetic Nephropathy in Human and Mouse Glomeruli. Diabetes 2013, 62, 299–308.
- Sha, Q.; Lyu, J.; Zhao, M.; Li, H.; Guo, M.; Sun, Q. Multi-Omics Analysis of Diabetic Nephropathy Reveals Potential New Mechanisms and Drug Targets. Front. Genet. 2020, 11, 616435.
- Eddy, S.; Mariani, L.H.; Kretzler, M. Integrated multi-omics approaches to improve classification of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 657–668.
- Woo, C.-Y.; Baek, J.Y.; Kim, A.-R.; Hong, C.H.; Yoon, J.E.; Kim, H.S.; Yoo, H.J.; Park, T.-S.; Kc, R.; Lee, K.-U.; et al. Inhibition of Ceramide Accumulation in Podocytes by Myriocin Prevents Diabetic Nephropathy. Diabetes Metab. J. 2020, 44, 581–591.
- Kalhorn, T.; Zager, R.A. Renal cortical ceramide patterns during ischemic and toxic injury: Assessments by HPLC-mass spectrometry. Am. J. Physiol. Content 1999, 277, F723–F733.
- Zager, R.A.; Conrad, S.; Lochhead, K.; Sweeney, E.A.; Igarashi, Y.; Burkhart, K. Altered sphingomyelinase and ceramide expression in the setting of ischemic and nephrotoxic acute renal failure. Kidney Int. 1998, 53, 573–582.
- Dupre, T.; Doll, M.A.; Shah, P.P.; Sharp, C.N.; Siow, D.; Megyesi, J.; Shayman, J.; Bielawska, A.; Bielawski, J.; Beverly, L.J.; et al. Inhibiting glucosylceramide synthase exacerbates cisplatin-induced acute kidney injury. J. Lipid Res. 2017, 58, 1439–1452.
- Zager, R.A.; Conrad, D.S.; Burkhart, K. Ceramide accumulation during oxidant renal tubular injury: Mechanisms and potential consequences. J. Am. Soc. Nephrol. 1998, 9, 1670–1680.
- Ueda, N.; Camargo, S.; Hong, X.; Basnakian, A.G.; Walker, P.D.; Shah, S.V. Role of Ceramide Synthase in Oxidant Injury to Renal Tubular Epithelial Cells. J. Am. Soc. Nephrol. 2001, 12, 2384–2391.
- Iwata, M.; Herrington, J.; Zager, R.A. Sphingosine: A mediator of acute renal tubular injury and subsequent cytoresistance. Proc. Natl. Acad. Sci. USA 1995, 92, 8970–8974.
- Cuvillier, O. Sphingosine in apoptosis signaling. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2002, 1585, 153–162.
- Woodcock, J. Sphingosine and ceramide signalling in apoptosis. IUBMB Life 2006, 58, 462–466.
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing Effects of ERK and JNK-p38 MAP Kinases on Apoptosis. Science 1995, 270, 1326–1331.
- Basnakian, A.G.; Ueda, N.; Hong, X.; Galitovsky, V.E.; Yin, X.; Shah, S.V. Ceramide synthase is essential for endonuclease-mediated death of renal tubular epithelial cells induced by hypoxia-reoxygenation. Am. J. Physiol. Physiol. 2005, 288, F308–F314.
- Siskind, L.J.; Mullen, T.D.; Rosales, K.R.; Clarke, C.J.; Hernandez-Corbacho, M.J.; Edinger, A.L.; Obeid, L. The BCL-2 Protein BAK Is Required for Long-chain Ceramide Generation during Apoptosis. J. Biol. Chem. 2010, 285, 11818–11826.
- Parra, V.; Eisner, V.; Chiong, M.; Criollo, A.; Moraga, F.; Garcia, A.; Härtel, S.; Jaimovich, E.; Zorzano, A.; Hidalgo, C.; et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc. Res. 2007, 77, 387–397.
- Yoo, T.-H.; Pedigo, C.E.; Guzman, J.; Correa-Medina, M.; Wei, C.; Villarreal, R.; Mitrofanova, A.; Leclercq, F.; Faul, C.; Li, J.; et al. Sphingomyelinase-Like Phosphodiesterase 3b Expression Levels Determine Podocyte Injury Phenotypes in Glomerular Disease. J. Am. Soc. Nephrol. 2014, 26, 133–147.
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701.
- Siskind, L.J.; Feinstein, L.; Yu, T.; Davis, J.S.; Jones, D.; Choi, J.; Zuckerman, J.E.; Tan, W.; Hill, R.B.; Hardwick, J.M.; et al. Anti-apoptotic Bcl-2 Family Proteins Disassemble Ceramide Channels. J. Biol. Chem. 2008, 283, 6622–6630.
- Kepp, O.; Rajalingam, K.; Kimmig, S.; Rudel, T. Bak and Bax are non-redundant during infection- and DNA damage-induced apoptosis. EMBO J. 2007, 26, 825–834.
- Zager, R.A.; Burkhart, K.M.; Johnson, A. Sphingomyelinase and Membrane Sphingomyelin Content. J. Am. Soc. Nephrol. 2000, 11, 894–902.
- Lan, T.; Liu, W.; Xie, X.; Xu, S.; Huang, K.; Peng, J.; Shen, X.; Liu, P.; Wang, L.; Xia, P.; et al. Sphingosine Kinase-1 Pathway Mediates High Glucose-Induced Fibronectin Expression in Glomerular Mesangial Cells. Mol. Endocrinol. 2011, 25, 2094–2105.
- Ishizawa, S.; Takahashi-Fujigasaki, J.; Kanazawa, Y.; Matoba, K.; Kawanami, D.; Yokota, T.; Iwamoto, T.; Tajima, N.; Manome, Y.; Utsunomiya, K. Sphingosine-1-phosphate induces differentiation of cultured renal tubular epithelial cells under Rho kinase activation via the S1P2 receptor. Clin. Exp. Nephrol. 2014, 18, 844–852.
- Jo, S.-K.; Bajwa, A.; Ye, H.; Vergis, A.L.; Awad, A.S.; Kharel, Y.; Lynch, K.R.; Okusa, M.D. Divergent roles of sphingosine kinases in kidney ischemia–reperfusion injury. Kidney Int. 2009, 75, 167–175.
- Park, S.W.; Kim, M.; Kim, M.; D’Agati, V.D.; Lee, H.T. Sphingosine kinase 1 protects against renal ischemia–reperfusion injury in mice by sphingosine-1-phosphate1 receptor activation. Kidney Int. 2011, 80, 1315–1327.
- Vidyasagar, A.; Wilson, N.A.; Djamali, A. Heat shock protein 27 (HSP27): Biomarker of disease and therapeutic target. Fibrogenesis Tissue Repair 2012, 5, 7.
- Kim, M.; Kim, M.; Kim, N.; D’Agati, V.D.; Sr, C.W.E.; Lee, H.T. Isoflurane mediates protection from renal ischemia-reperfusion injury via sphingosine kinase and sphingosine-1-phosphate-dependent pathways. Am. J. Physiol. Physiol. 2007, 293, F1827–F1835.
- Imeri, F.; Tanturovska, B.S.; Schwalm, S.; Saha, S.; Zeng-Brouwers, J.; Pavenstädt, H.; Pfeilschifter, J.; Schaefer, L.; Huwiler, A. Loss of sphingosine kinase 2 enhances Wilm’s tumor suppressor gene 1 and nephrin expression in podocytes and protects from streptozotocin-induced podocytopathy and albuminuria in mice. Matrix Biol. 2021, 98, 32–48.
- Bajwa, A.; Huang, L.; Kurmaeva, E.; Ye, H.; Dondeti, K.R.; Chroscicki, P.; Foley, L.S.; Balogun, Z.A.; Alexander, K.J.; Park, H.; et al. Sphingosine Kinase 2 Deficiency Attenuates Kidney Fibrosis via IFN-γ. J. Am. Soc. Nephrol. 2016, 28, 1145–1161.
- Yaghobian, D.; Don, A.; Yaghobian, S.; Chen, X.; Pollock, C.A.; Saad, S. Increased sphingosine 1-phosphate mediates inflammation and fibrosis in tubular injury in diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 2015, 43, 56–66.
- Park, S.W.; Kim, M.; Kim, J.Y.; Brown, K.M.; Haase, V.H.; D’Agati, V.D.; Lee, H.T. Proximal tubule sphingosine kinase-1 has a critical role in A1 adenosine receptor-mediated renal protection from ischemia. Kidney Int. 2012, 82, 878–891.
- Perry, H.M.; Huang, L.; Ye, H.; Liu, C.; Sung, S.-S.J.; Lynch, K.R.; Rosin, D.L.; Bajwa, A.; Okusa, M.D. Endothelial Sphingosine 1-Phosphate Receptor-1 Mediates Protection and Recovery from Acute Kidney Injury. J. Am. Soc. Nephrol. 2016, 27, 3383–3393.
- Bajwa, A.; Rosin, D.L.; Chroscicki, P.; Lee, S.; Dondeti, K.; Ye, H.; Kinsey, G.R.; Stevens, B.K.; Jobin, K.; Kenwood, B.M.; et al. Sphingosine 1-Phosphate Receptor-1 Enhances Mitochondrial Function and Reduces Cisplatin-Induced Tubule Injury. J. Am. Soc. Nephrol. 2014, 26, 908–925.
- Bajwa, A.; Jo, S.-K.; Ye, H.; Huang, L.; Dondeti, K.R.; Rosin, D.L.; Haase, V.H.; Macdonald, T.L.; Lynch, K.R.; Okusa, M.D. Activation of Sphingosine-1-Phosphate 1 Receptor in the Proximal Tubule Protects Against Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2010, 21, 955–965.
- Lai, L.-W.; Yong, K.-C.; Igarashi, S.; Lien, Y.-H. A sphingosine-1-phosphate type 1 receptor agonist inhibits the early T-cell transient following renal ischemia–reperfusion injury. Kidney Int. 2007, 71, 1223–1231.
- Lien, Y.-H.; Yong, K.-C.; Cho, C.; Igarashi, S.; Lai, L.-W. S1P1-selective agonist, SEW2871, ameliorates ischemic acute renal failure. Kidney Int. 2006, 69, 1601–1608.
- Kearney, P.M.; Whelton, M.; Reynolds, K.; Muntner, P.; Whelton, P.K.; He, J. Global burden of hypertension: Analysis of worldwide data. Lancet 2005, 365, 217–223.
- Vanhoutte, P.M. Endothelial dysfunction in hypertension. J. Hypertens Suppl. 1996, 14, S83–S93.
- Taddei, S.; Salvetti, A. Pathogenetic Factors in Hypertension Endothelial Factors. Clin. Exp. Hypertens. 1996, 18, 323–335.
- Di Pietro, P.; Carrizzo, A.; Sommella, E.; Oliveti, M.; Iacoviello, L.; Di Castelnuovo, A.; Acernese, F.; Damato, A.; De Lucia, M.; Merciai, F.; et al. Targeting the ASMase/S1P pathway protects from sortilin-evoked vascular damage in hypertension. J. Clin. Investig. 2022, 132.
- Pepe, G.; Cotugno, M.; Marracino, F.; Giova, S.; Capocci, L.; Forte, M.; Stanzione, R.; Bianchi, F.; Marchitti, S.; Di Pardo, A.; et al. Differential Expression of Sphingolipid Metabolizing Enzymes in Spontaneously Hypertensive Rats: A Possible Substrate for Susceptibility to Brain and Kidney Damage. Int. J. Mol. Sci. 2021, 22, 3796.