Sphingolipids (SLs) are lipids with complex structures which were named after their sphinx-like structure by Thudichium in 1870. Lipids are major components of membranes in all eukaryotic cells determining the structural and functional integrity of cells. SLs are one of the important structural components of plasma membranes in all eukaryotes, but they also act as bioactive signaling molecules with numerous cellular physiological functions that include cell adhesion, cell proliferation, cell migration, inflammatory response and apoptosis. Sphingolipids, which act as a bioactive signaling molecules, are involved in several cellular processes such as cell survival, proliferation, migration and apoptosis. An imbalance in the levels of sphingolipids can be lethal to cells. Abnormalities in the levels of sphingolipids are associated with several human diseases including kidney diseases. Several studies demonstrate that sphingolipids play an important role in maintaining proper renal function. Sphingolipids can alter the glomerular filtration barrier by affecting the functioning of podocytes, which are key cellular components of the glomerular filtration barrier.
1. Sphingolipid Biosynthesis and Catabolism
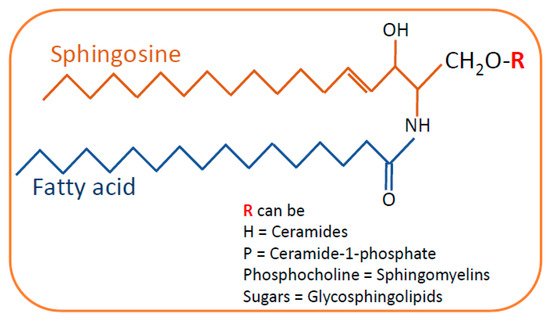
SLs are composed of a sphingosine backbone linked to a fatty acid with an amide bond. Based on the difference in the hydrophilic attachments (functional groups or substituents), SLs are categorized into ceramides, sphingomyelin and glycosphingolipids (
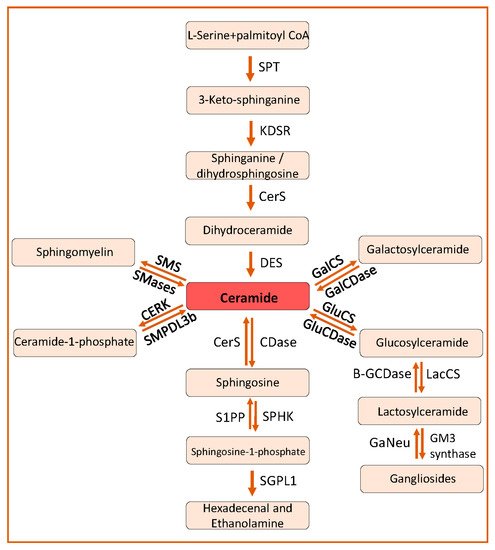
Figure 1). Ceramide is the central metabolite or substrate for the synthesis of complex SLs in the SL pathway. Ceramides can be synthesized by a de novo pathway and a salvage pathway (
Figure 2).
Figure 1. General structure of sphingolipids. Sphingosine is the backbone of the sphingolipid structure that is linked to fatty acids by an amide bond. Depending on the type of residue in the side chain, sphingolipids are categorized as ceramides, sphingomyelins and glycosphingolipids.
Figure 2. Sphingolipid pathway. The de novo sphingolipid metabolic pathway commences by the condensation of L-serine and palmitoyl-CoA to form 3-ketosphinganine, which ultimately is converted to ceramides by the action of KDSR, CerS and DES, respectively. Ceramides can also be synthesized from sphingomyelin by SMS, from glucosylceramide by GluCs and from galactosylceramide by GalCS. Ceramides can be phosphorylated to ceramide-1-phosphate by CERK and SMPDL3b can dephosphorylate ceramide-1-phosphate to ceramide. Ceramides can be deacylated to sphingosine by Cdases and sphingosine can be phosphorylated to form sphingosine-1-phosphate by SPHK. Sphingosine-1-phosphate can finally be broken down to hexadecanol and ethanolamine. SPT, serine palmitoyl transferase; KDSR, 3-ketosphinganine reductase; CerS, ceramide synthtase; DES, dihydroceramide desaturase; SMS, sphingomyelin synthetase, SMase, sphingomyelinase; CERK, ceramide kinase; SMPDL3b, Sphingomyelin Phosphodiesterase Acid Like 3b; CDase, ceramidase; SPHK, spingosine kinase; S1PP, spingosine-1-phosphate phosphatase; GluCS, glycosylceramide synthase; GalCS, galactosylceramide synthase; GluCDase, glycosylceramidase; GalCDase, galactosylceramidase; LacCs, lactosylceramide synthase; Β-GCDase, β-galactosidase; GaNeu, Ganglioside neuraminidase.
2. Sphingolipids in Kidney Diseases
The glomerulus of the kidney is an important structure which is essential to maintain the glomerular filtration barrier (GFB) that prevents leakage of proteins into the urine. Proper maintenance of this GFB is essential throughout life to prevent kidney injury and progression to kidney disease. Hence, understanding the factors contributing to kidney damage is important. In this review, we will focus on kidney diseases that are known to be caused due to imbalance in the levels of SLs and their association with podocyte injury and tubular damage.
2.1. Role of Sphingolipids in Podocyte Injury in Glomerular Diseases
The glomerular filtration barrier (GFB) is formed by podocytes, the glomerular basement membrane (GBM) and glomerular endothelial cells. Podocytes are terminally differentiated epithelial cells in the glomerulus. Foot processes of podocytes interdigitate with foot processes from adjacent podocytes, leaving filtration slits in between them, which are bridged by a structure called the slit diaphragm. Podocyte integrity, which mainly depends on the integrity of their actin cytoskeleton, is crucial for maintaining the proper function of the GFB [
32]. Podocyte dysfunction and loss of podocytes from the GFB due to cellular stress, genetic mutations, inflammation or lipotoxicity can lead to the leakage of proteins into urine, referred to as proteinuria. Podocyte injury and associated proteinuria is a hallmark of glomerular diseases [
33]. Podocyte injury contributes to the progression of several glomerular diseases, including diabetic kidney disease (DKD), focal segmental glomerulosclerosis (FSGS), Alport syndrome, IgA nephropathy and lupus nephritis (LN). In this section, we will focus on the glomerular diseases that are associated with imbalance in the levels of SLs and podocyte damage.
2.2. Role of Sphingolipids in Tubulointerstitial Fibrosis and Acute Kidney Injury
SLs have been shown to damage proximal tubular cells in many models of acute kidney injury (AKI), which include renal ischemia reperfusion injury (IRI), myoglobinuric AKI and cisplatin-induced AKI [
127,
136,
137]. The amount of ceramide levels present was found to be proportional to the extent of injury to proximal tubular cells [
127,
138], indicating that ceramides play a pivotal role in tubular cell damage possibly by inducing mitochondrial damage through reactive oxygen species (ROS) production and DNA damage [
9,
128]. Sphingosine from which ceramides can be synthesized mediates acute proximal tubular injury [
139]. Sphingosine-mediated tubular damage can be due to the inhibition of mitogen activated protein kinases (MAPKs) activity, ERK1 and ERK2 and activation of stress activated protein kinases (SAPKs), JNK and p38 [
140,
141], which is associated with apoptosis induced by a reduction in the expression of growth factors [
142]. Mass spectrometry analysis of SLs revealed that long and very long chain ceramide levels were increased in myoglobinuric-, cisplatin- and IRI-induced AKI causing injury to cells [
127,
136,
137] which can be attenuated by treating cells with fumonisin B1, a ceramide synthase inhibitor [
143]. Additionally, inhibiting ceramide synthesis by myriocin, a SPT complex inhibitor, prevented the cisplatin-mediated increase in ceramide levels and proximal tubular cell death by inducing pores in the mitochondrial outer membrane, mitochondrial outer membrane permeabilization and mitochondrial fragmentation [
144,
145,
146,
147,
148]. In support, the accumulation of ceramides due to increased sphingomyelinase activity was found to cause necrotic cell death of proximal tubular cells [
149], while the conversion of ceramides to glycosphingolipids reduced cell death in cisplatin-induced AKI [
137], indicating that ceramide accumulation plays a major role in proximal tubular cell injury.
The SPHK1 and S1P axis was shown to mediate the pathogenesis of tubular epithelial cells in DKD [
50,
150]. In
db/
db diabetic mice, it was shown that S1P-induced activation of Rho kinase-mediated fibrosis in renal tubular cells through S1PR2 disturbed the distribution of the adhesion molecule, E-cadherin, along the plasma membrane and of alpha-smooth muscle actin (α-SMA) in cytoplasm [
151]. SPHK2 knockout but not SPHK1 knockout mice exhibit aggravated injury after IRI, suggesting a protective role of SPHK2 in IRI [
152]. Contrary to this, another study showed that S1P synthesized by SPHK1 is important in the protection from IRI and that SPHK1 overexpression improves renal function via inducing the expression of heat shock protein 27 (HSP27) [
153] that acts as an antioxidant and inhibits apoptosis [
154]. Similarly, S1P and S1PR1 play a major role in renal injury after IRI as activation of S1P-dependent signaling and S1PR1 antagonism prevents isoflurane-mediated renal IRI and inflammation in both proximal tubular cells (HK2) and mice [
155]. Knockout of SPHK2 reduced renal fibrosis after AKI induced in folic acid or unilateral ischemia reperfusion by inducing the expression of IFN-gamma responsive genes such as
Cxcl9 and
Cxcl10 [
156]. Inhibiting S1P synthesis mediated by SPHK1 and SPHK2 was found to reduce tubulointerstitial renal inflammation and fibrosis in DKD and in human HK2 cells by reducing the expression of fibronectin, collagen IV and macrophage chemoattractant protein 1 (MCP1) [
157]. From these studies, it can be concluded that the SPHK and S1P axis plays an important role in the protection of proximal tubular cells from renal IRI [
158,
159].
S1P receptors also play a major role in tubular cell damage. Loss of S1PR1 in proximal tubular cells exacerbates IRI [
55], which in turn can be inhibited by increasing S1P synthesis, S1PR1 receptor agonism and SPHK gene delivery [
55,
155]. Increased apoptosis is observed in S1PR1 knockout mice treated with cisplatin and activation of S1PR1 by FTY720 was shown to rescue cisplatin-induced AKI by stabilizing the mitochondrial function by decreasing cytochrome c release and regulation of BCL-2 proteins [
160]. SEW2871 also prevented apoptosis and attenuated IRI whereas VPC4416, a S1PR1 antagonist, aggravated IRI and inflammation associated with tubular injury induced by LPS [
55,
161,
162,
163]. Interestingly, S1PR2 antagonists or mice that lack S1PR2 are protected from renal IRI and SPHK antagonism or S1PR2 agonism exacerbates renal IRI [
153,
155], indicating that S1P signaling mediated by S1PR1 protects tubular cells, whereas S1P signaling mediated by S1PR2 causes tubular cell injury. Though proximal tubular cells are highly susceptible cells to injury during renal IRI, renal endothelial cell damage is also observed. Loss of S1PR1 in renal endothelial cells prevented the recovery from IRI and even aggravated renal injury, inflammation and renal fibrosis [
159]. S1PR1 was shown to suppress endothelial activation of leukocyte adhesion molecule expression and inflammation [
159].
Endothelial dysfunction is a major contributor for hypertension that can lead to cardiovascular and kidney diseases [
164,
165,
166]. Plasma sortilin levels were increased in hypertensive patients with endothelial dysfunction along with an increase in ASMase activity, plasma S1P and soluble NADPH oxidase 2 (NOX2)-derived peptide [
167]. Sortulin induces endothelial dysfunction of NOX2 activation, increasing oxidative stress and altering SL metabolism. Sortulin levels are associated with altered SL metabolism and oxidative stress in patients with arterial hypertension [
167]. LC-MS analysis of sortilin-treated endothelial cells showed a decrease in ceramides (C16–C24) with an increase in S1P [
167]. S1P was shown to play a significant role in hypertension-mediated kidney damage [
167,
168,
169]. Sortilin-induced endothelial dysfunction was prevented by knockdown of either ASMase or SPHK1 [
167]. Sortilin was found to induce vascular oxidative stress by promoting Rac1-mediated activation of NOX2 that was prevented by inhibition of S1PR3-mediated signaling [
167]. Another study showed that Nogo-B (ER membrane protein and reticulon-4 family protein) regulates hypertension through endothelial nitric oxide synthase (eNOS) pathway [
169]. Endothelial S1P-S1PR1 signaling is important for eNOS pathway activation [
170]. LC-MS analysis of SLs in Nogo-B-deficient primary endothelial cells isolated from mouse lung showed an increase in sphingosine and S1P [
169]. Nogo-B inhibits SPT expression thereby controlling the S1P production and its signaling mediated by S1PR1, thus restoring the endothelial dysfunction [
169]. Recently, it was shown that expression of genes related to SL metabolism were altered in renal tissues of hypertensive rats when compared to normotensive rats [
168]. Expression of S1PR1 was increased in both spontaneously hypertensive rats that are stroke resistant (SHSR) and spontaneously hypertensive rats that are stroke prone (SHSP) when compared to normotensive Wistar Kyoto rats [
168]. SHSR rats were characterized by reduced expression of CerS2 mRNA in kidneys, whereas SHSP rats were characterized by reduced protein expression of SPTLC2 in kidneys [
168].
Glycosphingolipid globotriaosylceramide (Gb3), which is expressed in renal proximal tubular epithelia, was shown to influence the reabsorption of proteins and albumin in proximal tubules. Inhibition of Gb3 synthesis blocks the reabsorption of proteins and protects from tubular damage [
171]. Lyso-Gb3 which induces the epithelial–mesenchymal transition (EMT) response in HK2 cells in a TGF-β-dependent pathway increases the expression of extracellular matrix (ECM) components, thus contributing to disease progression, which can be prevented by inhibiting TGF-β receptor [
172].
Taken together, studies in tubular cells support the idea that SLs and tubular cell-specific targeting of genes regulating the SL pathway may represent a therapeutic option to prevent tubular injury and to attenuate the effects of renal IRI.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23084244