This sarcoma subtype is likely derived from unique progenitor cells with similar genetic backgrounds belonging to endothelial, mesodermal, epithelial, and nerve cells [
], but most studies suggest that mesenchymal stem cells (MSCs) are the main precursor cells [
]. Ewing sarcoma belongs to the Ewing sarcoma family tumors (ESFT), which in turn belongs to the FET (FUS-EWSR1-TAF15) group of sarcomas and leukemias. The tumor group comprises more than 20 different tumor entities with ES as one of the most common types [
]. These tumors have similar non-random chromosomal translocations with one of the three genes, such as EWSR1, FUS, and TAF15, as 5′ partners and a large group of DNA binding transcription factor encoding genes as 3′ partners. Of note, these fusion proteins encode ETS biding factors that bind to similar DNA sequences. For ES the t(11;22)(q24;q12) translocation is present in 85–90% of tumors and
). The t(11;22) (q24; q12) translocation is found in 85–90% of ES tumors, and the EWSR1 and FLI1 gene fusion results in a fusion protein (EWSR1/FLI1). Further, the t(21;22)(q22;q12), t(7;22)(q22;q12), t(7;22)(q22;q12), t(17;22)(q12;q12), t(2;22)(q33;q12) and others are less common translocations that result in the development of
].
The two most common fusion proteins (
EWSR1/FLI1 and
EWSR1/ERG) [
17] are involved in several cell signaling and regulatory pathways (Graphical Abstract). These fusion proteins usually act as transcription factors. For instance, Boulay et al. [
22] identified a chromatin-binding factor (BAF) that interacts with EWSR1/FLI1 to activate gene expression in ES tumor cells and phenotypical changes. On the other hand, oncoprotein fusion can induce target genes via a partnership with GGAA microsatellites, as active enhancers [
23] or by binding to RNA. Important discoveries in recent years have shown that major fractions of the ES fusion proteins bind to the SWI/SNF chromatin remodeling complex in tumor cells and that leads to the deregulation of gene expression, such as IGF-1 signaling [
24] and epigenetic programming [
15,
16] towards retaining mesenchymal stem cell plasticity [
25]. Blockade of these fusion proteins may be ideal for therapeutic targets. The feasibility of
EWSR1/FLI1 targeting for ES therapy has been shown preclinically using chemical inhibitors [
26,
27,
28] and siRNA technology (Bertrand et al. [
29] Gauthier et al. [
30] and Cervera et al. [
31]) and via chemical targeting of
EWSR1/FLI1 oncogenic fusion with chemical inhibitors [
32]. Despite affecting Ewing sarcoma cell viability, multiple reports reveal induction of resistance factors contributing to the tumor’s survival and therapy escape [
33,
34,
35]. Given this problem, it seems prudent to consider the combination of inhibitors against
EWSR1/FLI1 or
EWSR1/ERG with additional targeted therapies [
36,
37] that might overcome tumor cells’ resistance to the monotherapy and might demonstrate a less adverse effect due to dose reduction. One such drug is YK-4-279, a drug candidate active in downregulating transcription of EWS/FLI1 [
38]. At the same time, YK-4-279 has been shown to inhibit ERG and ETV1 transcription in prostate cancer cells [
39], therefore, it might also be active against Ewing sarcoma cells with commonly and less commonly presented fusion in the tumors with Ewing sarcoma.
3.1. Targeting of ES Pressure on Adhesion, Migration, and Invasion
Like most cancer types, the prognosis for ES patients with localized disease is much better than for patients with metastatic disease [
40]. A major obstacle in the battle against metastatic disease continues to be an insufficient understanding of underlying processes; specifically, what processes, such as metabolic and others, drive cell adhesion, migration, and invasion during metastases.
Adhesion proteins, such as E-cadherin are regarded as tumor suppressors. The low level of epithelial proteins is associated with spheroid formation and migration [
41]. The migration of cancer cells to new niches is a fundamental process underlying metastasis [
42]. There are two main types of cancer migration; mesenchymal and amoeboid; each is dependent on complex intracellular signaling that governs the actin cytoskeleton. Actin is involved in several pathways, including Rho/Rac GTPases and the Hippo-pathway, which culminate in the transcriptional regulation of cytoskeletal and growth-promoting genes, respectively. The mesenchymal cell migration of sarcomas can involve both single cells and cells in chains [
43]. For successful metastasis, migration is followed by invasion into secondary locations [
44].
Cadherins are vital for the formation of cell-cell contacts. During tumor progression, E-cadherin expression is lost, which permits epithelial to mesenchymal transition, anchorage-independent growth, and spheroid formation [
45]. Studies have shown that loss of E-cadherin promotes resistance to treatment [
40] and acquisition of a mesenchymal-like phenotype in vitro. Ex vivo studies have shown that the increased expression of E-cadherin is associated with improved clinical outcomes in several types of sarcomas [
46]. Thus, E-cadherin upregulation in ES cells is mediated by epigenetics [
47], or by small molecules like MIL327 [
40] or RNA interference to metalloproteinase type 9 (MMP9) [
48].
Besides induction of the epithelial to mesenchymal transition (EMT)-developed cell signaling, ES metastasis requires cell migration and blood vessel invasion [
49]. The Rho-associated kinases, ROCK1 and ROCK2 have been implemented in the regulation of metastases using various in vitro and in vivo models. Data by Roberto et al. [
49] showed a positive correlation between miR-139-5p and ROCK1, where restoration of miR-139-5p impaired ES migration and invasion. Another approach is the application of ROCK2 inhibitors, such as SR3677 and hydroxyfasudil, in SK-ES-1 cells [
42] to demonstrate an opportunity for simultaneous targeting of heterogenic ES cells.
The TGF-β and PDGF pathways play important roles in ES plasticity and tumor progression. The TGF-β co-receptor endoglin is routinely expressed by malignant cells, and it is associated with the upregulation of bone morphogenetic protein, integrin, focal adhesion kinase, and phosphoinositide-3-kinase signaling, which together work in concert to maintain tumor cell plasticity [
43,
83]. Like TGF-β, the platelet-derived growth factor (PDGF) pathway aids in maintaining a cancer stem cell-like phenotype in ES, but more notably, is involved in ES tumor neovascularization [
84]. Importantly PDGF ligands and/or receptors are frequently upregulated in ES [
66,
85] and their expression correlates with the activity of the
EWSR1/FLI1 fusion [
86] in sarcoma tissues. Overall, the pathways involved in ES cellular invasion and migration are complex, and future work is needed to design an effective multitargeted approach against ES tumor cells [
87].
These genes play a key role in several tumor processes for ES, especially in adhesion, migration, and invasion (
Table 1). The regulation of these processes in a tumor is the first step toward the formation of a lesion secondary tumor growth-metastasis from a localized tumor. Metastasis is associated with poor clinical outcomes for patients, while the treatment of localized tumors currently has a relatively high success rate, it could be as high as 70% [
18]. Therefore, these genes as targets for therapy are especially vital.
Table 1. Targeting molecules in ES pathogenesis.
|
Targetable Molecules
|
Main Pathways
|
Tumor Effects
|
|
CD99
|
IGF-1R and RAS-Rac1 signaling
|
Induces caspase-independent cell death, endocytosis, cell aggregation, micropinocytosis, cell adhesion, migration, invasion, metastasis, differentiation
|
|
GDF6
|
GDF6 prodomain signaling pathway
|
Cell proliferation, tumor growth, differentiation, apoptosis
|
|
E-cadherin
|
MAPK Pathway
|
Anchorage-independent growth and spheroid formation, cell-cell adhesion
|
|
Endoglin
|
TGFβ signaling
|
Tumor cell plasticity, patient survival, invasion, anchorage-independent growth, progression of aggressive tumors
|
|
EZH2
|
Epigenetic
|
Cell differentiation, phenotypic heterogeneity, self-renewal
|
|
GLI1
|
Sonic Hedgehog (SHH) pathway
|
Cell proliferation, cell cycle control, apoptosis, cell viability, metastasis, invasion, migration, clonogenicity
|
|
PDGF family members
|
PDGF pathway
|
Self-renewal, invasion, chemotherapy resistance, primary tumor growth, metastasis, drug resistance, poor clinical outcome
|
|
ROCK2
|
RhoA-ROCK pathway
|
Migration, invasion, proliferation, clonogenic capacity, tumor growth
|
|
YAP proteins
|
YAP/TAZ pathway, Hippo signaling, WNT/β-catenin signaling
|
Migration, cell proliferation, metastasis, anchorage-independent colony formation
|
|
PAPP-A
|
IGF signaling
|
Cell proliferation, migration, cell survival, tumor growth, invasion, metastasis
|
|
PARP family
|
DNA repair, replication
|
Apoptosis
|
|
TRAIL
|
TRAIL-pathway
|
Induces caspase-independent cell death, apoptosis
|
|
ATR/CHK1
|
ATR-CHK1 pathway
|
Cell cycle regulation, cell cycle arrest
|
|
LDH
|
aerobic glycolysis
|
Cell proliferation, apoptosis, tumor growth, cell survival
|
|
PHGDH
|
Serine synthesis
|
Cell proliferation
|
|
lncRNA SOX2
|
WNT/β-catenin signaling
|
Cell proliferation, invasion, apoptosis, tumor growth
|
|
lncRNA TUG1
|
TUG-miR-145-5p-TRPC6 pathway
|
Cell proliferation, migration, invasion
|
3.2. Targeting of Ewing Sarcoma Cells with a Focus on Proliferation, Cell Differentiation, and Cell Survival
Metastatic progression requires the migration and invasion of cancer cells to reach distant tissues. However, for malignant neoplasms to spread beyond their primary tumor, they must adapt and thrive in a new niche. In general, cancer cells are characterized by unlimited time spent on cell division coupled with high survivability [
88]. The mechanism for preserving these cells in a poorly differentiated state is elaborate. Mutations and epigenetic changes trigger unregulated mitotic cycles, allowing cells to become insensitive to growth-inhibitory signals, and capable of evading programmed cell death. Cell cycle progression and cell division, cell death, and cellular senescence determine cell proliferation in a broad sense.
The expression of the ES fusion gene alters the expression of over 500 downstream targets, which collectively block differentiation and drive proliferation [
89]. For example,
EWSR1/FLI1 regulates several genes, including IGF-1, Homeobox protein Nkx (NKX2), T-LAK cell-originated protein kinase (TOPK), SRY-Box Transcription Factor 2 (SOX2), and Enhancer Of ZesteHomolog2 (EZH2) [
90]. It is worth noting that EWS-FLI1 binds to the
EZH2 promoter to activate embryonic tumor stem cell growth and metastatic spread [
91]. These targets can be used in the treatment of Ewing sarcoma.
It is well-known that several cancers [
99,
100] heavily rely upon glycolysis rather than oxidative metabolism since it provides metabolic plasticity to fuel tumor heterogeneity [
101,
102]. Ewing sarcoma is one tumor type [
103] where a predominant fusion protein,
EWS/FLI1, regulates glucose consumption as well as gene expression of glycolytic enzymes, such as lactate dehydrogenase (LDH) [
32]. It has been shown recently that depletion of lactate dehydrogenase-A (LDHA) inhibits proliferation of ES cells and induces apoptosis, impacting tumor cell viability both in vitro and in vivo.
Additionally, for various cancers, a growing number of important regulators are being discovered within one group of molecules: long non-coding RNAs (lncRNAs) [
104]. LncRNAs often increase cancer cell survival, proliferation, colony formation, migration, and invasion [
104,
105,
106]. Their elevated expression contributes to the progression of the sarcoma. It was described, for example, for lncRNA taurine upregulated gene 1 (TUG1) [
106] and lncRNA SOX2 [
105]. Many of the biological regulatory mechanisms of lncRNAs in Ewing sarcoma are still elusive. Nevertheless, they have been shown to often act as competing endogenous RNAs to regulate other genes expression. In principle, the knockdown of lncRNAs or the selection of inhibitory proteins might help to suppress ES growth [
105,
106]. These approaches might be potential therapeutic options for treating Ewing sarcoma.
In summary, cell proliferation, differentiation, and survival are important, first of all, in the formation of a tumor in a primary or secondary lesion. These properties of cells are related to some extent to other tumor characteristics (Figure 1), for example, deregulated cellular energetics (Table 1). To regulate this group of genes and their products, EWSR1/FLI1 usually acts as a transcription factor and binds with RNA.
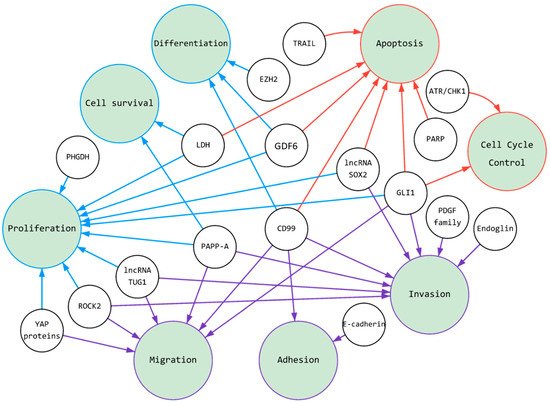
Figure 1. Schematic illustration of cancer pathways under control of ES’s fusion proteins: The figure summarizes the researchers' review of research active molecules over the past 10 years. The various domain of ES-produced fusion oncoproteins required for the activation of gene expression, and their products, such as RNA or proteins regulate cellular proliferation, apoptosis and migration, for example. All together with induced cell signaling cells gain oncogenic traits and transformation. Small circles are active molecules, large circles are cellular processes (see the hallmark of cancer). Colors in the figure are correlated to organizational divisions here for ease of perception. There are three colored clusters: purple, blue and red.
3.3. Targeting of ES: Induction of Apoptosis and Cell Cycle Arrest
The development of fusion event inhibitors to suppress the progression of Ewing sarcoma requires the understanding of multiple cellular processes which became active in the tumor cells. Since kinases are integral to tumor maintenance, intervention directed toward these family members is promising. Both apoptosis and cell division utilize several different kinases, such as ATR and CHK1.
ES cells exhibit increased levels of endogenous DNA replicative stress and are sensitive to inhibitors of ribonucleotide reductase (RNR), an enzyme that limits the rate of deoxyribonucleotide synthesis. ES cells are also dependent on the ataxia telangiectasia, the rad3-related protein (ATR), and the checkpoint kinase 1 (CHK1) pathway, which play key roles in orchestrating the cellular response to DNA replication stress for survival [
107,
108]. ES tumors are sensitive both in vitro and in vivo to ATR and CHK1 inhibitors as separate agents and in combination with other drugs [
107,
108,
109,
110,
111]. The ATR-CHK1 pathway, when activated by DNA replication stress, orchestrates a multifaceted response that arrests cell cycle progression, suppresses the origin of replication, stabilizes replication forks, and promotes fork repair and restart [
112].
Additionally, one of the enzymes regulating the work of the hereditary apparatus of the cell is Poly (ADP-ribose) polymerase (PARP), which is involved in the processes of DNA repair, maintaining the genetic stability of the cell and its programmed death, and as Ewing sarcoma cell lines are frequently defective in DNA break repair, they are susceptible to PARP inhibition [
115,
116,
117]. Inhibition of PARP showed the effectiveness of this approach if cytostatic drugs were used, the effect of which was intensified [
118].
Growth and differentiation factor 6 (GDF6), also known as bone morphogenetic protein 13 (BMP13), belongs to the TGF-superfamily’s BMP family. GDF6 has become an attractive target since its binding to its own receptor, such as CD99 [
41]. GDF6 is vital to embryogenesis, particularly to the development of the neural and skeletal systems, and mutations in GDF6 are associated with abnormalities of the skeleton, the eyes [
124], and other organs [
125]. GDF6 is highly expressed in ES tumors and cell lines compared to mesenchymal stem cells and cells from other sarcoma subtypes.
The anticancer properties of multiple ES-based therapeutic approaches have been extensively studied, often for their antiproliferative effects. The induction of apoptosis has been reported for several cancers, including Ewing sarcoma. However, ES cells are prone to developing treatment resistance, which contributes to disease recurrence [
126]. Current ES-therapeutic strategies are promising, and the development of new therapeutics and combinations thereof with greater antitumoral properties has been proposed.
Previously, TRAIL, a member of the tumor necrosis factor (TNF) ligand superfamily (TNFLSF), has been found to induce apoptosis in cancer cells while sparing normal cells [
129,
130,
131]. Multiple in vitro studies have shown that TRAIL and other death receptor agonists [
132,
133] are effective against sarcoma cell lines [
131,
134] with ES cell lines showing the greatest sensitivity [
135]. Ubiquitin-specific protease 6 (USP6) may also contribute to the sensitization of ES cells to exogenous IFNs [
136]. Henrich and colleagues [
136] speculate that this negative feedback loop involves USP6, which serves to amplify Interferon Gamma (IFN)-mediated sensitivity to TRAIL. Indisputably, the molecular mechanism of TRAIL sensitivity warrants additional investigation to clarify the molecular basis for drug synergy against ES.
The metabolism of Ewing sarcoma involves many genes and metabolic pathways that may be potential targets for therapy (
Table 1). The effect on some agents can lead to a static effect, and the activation or silencing of others can be expressed as a bright antitumor effect and lead to apoptosis. Some experimental approaches to stimulating cell cycle arrest [
137,
138,
139] or apoptosis [
140,
141] in Ewing sarcoma cells showed effectiveness in preclinical settings, and therefore, might hold great promises in future clinical testing.