Ageing is a complex and unavoidable process that can be defined as the functional decline after a period of maturity. In this respect, maturity is understood as the endpoint of development and the condition of maximal functional performance capability. The nucleolus organizes around the sites of transcription by RNA polymerase I (RNA Pol I). rDNA transcription by this enzyme is the key step of ribosome biogenesis and most of the assembly and maturation processes of the ribosome occur co-transcriptionally. Therefore, disturbances in rRNA transcription and processing translate to ribosomal malfunction. Nucleolar malfunction has recently been described in the classical progeria of childhood, Hutchinson–Gilford syndrome (HGPS), which is characterized by severe signs of premature aging, including atherosclerosis, alopecia, and osteoporosis. A deregulated ribosomal biogenesis with enlarged nucleoli is not only characteristic for HGPS patients, but it is also found in the fibroblasts of “normal” aging individuals. Cockayne syndrome (CS) is also characterized by signs of premature aging, including the loss of subcutaneous fat, alopecia, and cataracts. It has been shown that all genes in which a mutation causes CS, are involved in rDNA transcription by RNA Pol I.
- nucleolus
- aging
- RNA Pol I
- ribosome
- loss of proteostasis
- neurodegeneration
1. Aging and the Nucleolus
2. Nucleolar Size as a Hallmark of Aging
3. Cockayne Syndrome (CS)
4. Loss of RNA Pol I Transcription Leads to Mitochondrial Dysfunction
5. Loss of Proteostasis in CS
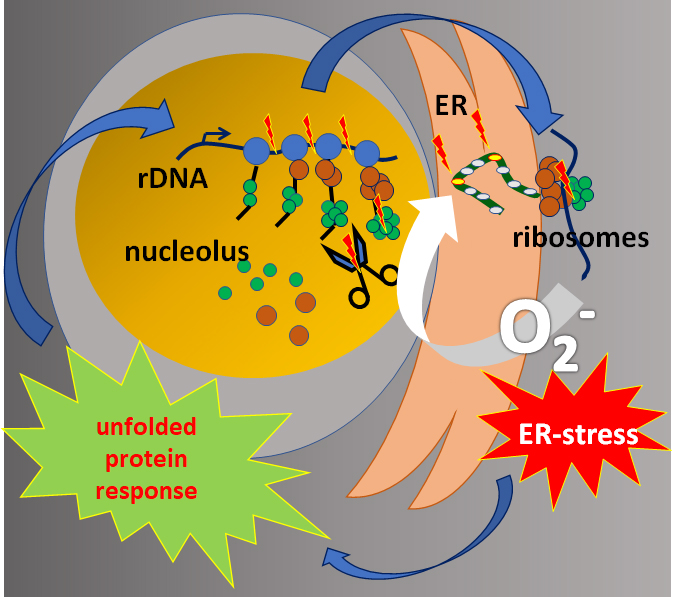
This entry is adapted from the peer-reviewed paper 10.3390/cells8060534
References
- Bassler, J.; Hurt, E. Eukaryotic Ribosome Assembly. Annu. Rev. Biochem. 2018, 88.
- Andersen, J.S.; Lyon, C.E.; Fox, A.H.; Leung, A.K.; Lam, Y.W.; Steen, H.; Mann, M.; Lamond, A.I. Directed proteomic analysis of the human nucleolus. Curr. Biol. 2002, 12, 1–11.
- Bensaddek, D.; Nicolas, A.; Lamond, A.I. Quantitative proteomic analysis of the human nucleolus. In The Nucleolus; Humana Press: New York, NY, USA, 2016.
- Kennedy, B.K.; Gotta, M.; Sinclair, D.A.; Mills, K.; McNabb, D.S.; Murthy, M.; Pak, S.M.; Laroche, T.; Gasser, S.M.; Guarente, L. Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell 1997, 89, 381–391.
- Sinclair, D.A.; Guarente, L. Extrachromosomal rDNA circles—A cause of aging in yeast. Cell 1997, 91, 1033–1042.
- Penzo, M.; Montanaro, L.; Trere, D.; Derenzini, M. The Ribosome Biogenesis-Cancer Connection. Cells 2019, 8, 55.
- Tiku, V.; Antebi, A. Nucleolar Function in Lifespan Regulation. Trends Cell Biol. 2018, 28, 662–672.
- Antikainen, H.; Driscoll, M.; Haspel, G.; Dobrowolski, R. TOR-mediated regulation of metabolism in aging. Aging Cell 2017, 16, 1219–1233.
- Finkel, T. The metabolic regulation of aging. Nat. Med. 2015, 21, 1416–1423.
- Lopez-Otin, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821.
- Tiku, V.; Jain, C.; Raz, Y.; Nakamura, S.; Heestand, B.; Liu, W.; Spath, M.; Suchiman, H.E.D.; Muller, R.U.; Slagboom, P.E.; et al. Small nucleoli are a cellular hallmark of longevity. Nat. Commun. 2017, 8, 16083.
- Frank, D.J.; Roth, M.B. ncl-1 is required for the regulation of cell size and ribosomal RNA synthesis in Caenorhabditis elegans. J. Cell Biol. 1998, 140, 1321–1329.
- Hedgecock, E.M.; Herman, R.K. The ncl-1 gene and genetic mosaics of Caenorhabditis elegans. Genetics 1995, 141, 989–1006.
- Buchwalter, A.; Hetzer, M.W. Nucleolar expansion and elevated protein translation in premature aging. Nat. Commun. 2017, 8, 328.
- Ferri, D.; Orioli, D.; Botta, E. Heterogeneity and overlaps in nucleotide excision repair (NER) disorders. Clin. Genet. 2019.
- Nance, M.A.; Berry, S.A. Cockayne syndrome: Review of 140 cases. Am. J. Med Genet. 1992, 42, 68–84.
- Laugel, V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013, 134, 161–170.
- Theil, A.F.; Hoeijmakers, J.H.; Vermeulen, W. TTDA: Big impact of a small protein. Exp. Cell Res. 2014, 329, 61–68.
- Brooks, P.J. Blinded by the UV light: how the focus on transcription-coupled NER has distracted from understanding the mechanisms of Cockayne syndrome neurologic disease. DNA Repair 2013, 12, 656–671.
- Bohr, V.; Anson, R.M.; Mazur, S.; Dianov, G. Oxidative DNA damage processing and changes with aging. Toxicol. Lett. 1998, 102–103, 47–52.
- D’Errico, M.; Pascucci, B.; Iorio, E.; Van Houten, B.; Dogliotti, E. The role of CSA and CSB protein in the oxidative stress response. Mech. Ageing Dev. 2013, 134, 261–269.
- Sampath, H. Oxidative DNA damage in disease--insights gained from base excision repair glycosylase-deficient mouse models. Environ. Mol. Mutagenesis 2014, 55, 689–703.
- Khobta, A.; Epe, B. Repair of oxidatively generated DNA damage in Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 253–260.
- Tamura, D.; DiGiovanna, J.J.; Khan, S.G.; Kraemer, K.H. Living with xeroderma pigmentosum: comprehensive photoprotection for highly photosensitive patients. Photodermatol. Photoimmunol. Photomed. 2014, 30, 146–152.
- Horibata, K.; Iwamoto, Y.; Kuraoka, I.; Jaspers, N.G.; Kurimasa, A.; Oshimura, M.; Ichihashi, M.; Tanaka, K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 15410–15415.
- Nardo, T.; Oneda, R.; Spivak, G.; Vaz, B.; Mortier, L.; Thomas, P.; Orioli, D.; Laugel, V.; Stary, A.; Hanawalt, P.C.; et al. A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc. Natl. Acad. Sci. USA 2009, 106, 6209–6214.
- Itoh, T.; Fujiwara, Y.; Ono, T.; Yamaizumi, M. UVs syndrome, a new general category of photosensitive disorder with defective DNA repair, is distinct from xeroderma pigmentosum variant and rodent complementation group I. Am. J. Hum. Genet. 1995, 56, 1267–1276.
- Soltys, D.T.; Rocha, C.R.; Lerner, L.K.; de Souza, T.A.; Munford, V.; Cabral, F.; Nardo, T.; Stefanini, M.; Sarasin, A.; Cabral-Neto, J.B.; et al. Novel XPG (ERCC5) mutations affect DNA repair and cell survival after ultraviolet but not oxidative stress. Hum. Mutat. 2013, 34, 481–489.
- Yu, Y.; Cui, Y.; Niedernhofer, L.J.; Wang, Y. Occurrence, Biological Consequences, and Human Health Relevance of Oxidative Stress-Induced DNA Damage. Chem. Res. Toxicol. 2016, 29, 2008–2039.
- Alupei, M.C.; Maity, P.; Esser, P.R.; Krikki, I.; Tuorto, F.; Parlato, R.; Penzo, M.; Schelling, A.; Laugel, V.; Montanaro, L.; et al. Loss of Proteostasis Is a Pathomechanism in Cockayne Syndrome. Cell Rep. 2018, 23, 1612–1619.
- Iben, S.; Tschochner, H.; Bier, M.; Hoogstraten, D.; Hozak, P.; Egly, J.M.; Grummt, I. TFIIH plays an essential role in RNA polymerase I transcription. Cell 2002, 109, 297–306.
- Hoogstraten, D.; Nigg, A.L.; Heath, H.; Mullenders, L.H.; van Driel, R.; Hoeijmakers, J.H.; Vermeulen, W.; Houtsmuller, A.B. Rapid switching of TFIIH between RNA polymerase I and II transcription and DNA repair in vivo. Mol. Cell 2002, 10, 1163–1174.
- Bradsher, J.; Auriol, J.; Proietti de Santis, L.; Iben, S.; Vonesch, J.L.; Grummt, I.; Egly, J.M. CSB is a component of RNA pol I transcription. Mol. Cell 2002, 10, 819–829.
- Schmitz, K.M.; Schmitt, N.; Hoffmann-Rohrer, U.; Schafer, A.; Grummt, I.; Mayer, C. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol. Cell 2009, 33, 344–353.
- Koch, S.; Garcia Gonzalez, O.; Assfalg, R.; Schelling, A.; Schafer, P.; Scharffetter-Kochanek, K.; Iben, S. Cockayne syndrome protein A is a transcription factor of RNA polymerase I and stimulates ribosomal biogenesis and growth. Cell Cycle 2014, 13, 2029–2037.
- Scheibye-Knudsen, M.; Tseng, A.; Borch Jensen, M.; Scheibye-Alsing, K.; Fang, E.F.; Iyama, T.; Bharti, S.K.; Marosi, K.; Froetscher, L.; Kassahun, H.; et al. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc. Natl. Acad. Sci. USA 2016, 113, 12502–12507.
- Azpurua, J.; Ke, Z.; Chen, I.X.; Zhang, Q.; Ermolenko, D.N.; Zhang, Z.D.; Gorbunova, V.; Seluanov, A. Naked mole-rat has increased translational fidelity compared with the mouse, as well as a unique 28S ribosomal RNA cleavage. Proc. Natl. Acad. Sci. USA 2013, 110, 17350–17355.