Biosimilars have come of age over the past 17 years, with 84 approvals in the EU and 35 in the US, representing almost 90% of the world market. While the acceptance of biosimilars in the US is catching up with that in the EU, the cost benefits remain elusive due to the high development barrier and complex distribution system involved, mainly in the US. In the EU, the cost of biosimilars has already dropped 70% or more, and interchangeability is a routine in some European jurisdictions, unlike in the US, where a separate regulatory approval is required. This paper projects significant changes coming in the US and EU’s biosimilars approval requirements that will impact the approval procedures in the rest of the world, leading to dramatic changes in the cost of biosimilars to patients. This perspective is based on the author’s first-hand experience to secure FDA approvals of biosimilars and an extensive analysis of the rationality of testing to demonstrate biosimilarity. Multiple citizen petitions by the author and meetings with the FDA may have prompted the recent announcement by the FDA to award a $5 million research grant to scientists to develop novel testing models to establish biosimilarity, including modifying the interchangeability protocols. Soon, demonstration of biosimilarity will not require animal testing and, in most cases, clinical efficacy testing; over time, the clinical pharmacology testing will be reduced as the regulatory agencies develop more confidence in the safety and efficacy of biosimilars. Biosimilars have come of age; now it is the turn of the developers to grow up, and one way to show this is to challenge the current regulatory guidelines but only on scientific grounds to seek more concessions, for which both FDA and EMA are ready
1. Introduction
The first biosimilar, Sandoz’s Omnitrope [
1] (human growth hormone), was approved in 2006 by both the FDA and EMA; it was approved by the FDA under the 505(b)(2) generic product legislation and by the EU as a biosimilar. Later, Sandoz received the first licensing of Zarzio/Zarxio (filgrastim) in 2015; this was the first biosimilar product approved under the 351(k) BLA legislation in the US [
2]. Many firsts were to follow in the US: the first mAb (bevacizumab), the first pegylated cytokine (pegfilgrastim), the first ophthalmic biosimilar (ranibizumab), and the first two interchangeable biosimilars (insulin glargine and adalimumab) [
3]. The US also approved the first biosimilars without clinical efficacy testing (filgrastim, pegfilgrastim, and erythropoietin alfa).
In 2021, the FDA approved four products, had three launches, and accumulated more than 100 development plans for biosimilar enrollment. The EMA approved seven products in 2021, with 16 more being under review [
4,
5]. Being under review and having plan enrollment are two different metrics. Still, more companies are heading to the FDA, as filgrastim, trastuzumab, bevacizumab, and rituximab biosimilars capture over half of their respective markets; infliximab biosimilars, which had a low penetration at first, have now captured more than a quarter of the market in the US. Amgen reported selling USD 2 billion of biosimilars in the first nine months of 2021 [
6]. In addition, US prescribers are becoming more used to biosimilars, as shown by the quick uptake of insulin glargine products. The biosimilars for adalimumab will enter the market in 2023, and their high adoption is expected.
While most biosimilars have rigidly followed the WAC pricing [
7] of 3–30% below the reference product in the US, we can anticipate significant price drops in the future. Since the US market represents 40% of the world market [
8], price drops in the US are pivotal in presenting the overall benefits of biosimilars. In the EU, the pricing of biosimilars is a regional issue, presenting a range of 30 to 70% of market share and price drops of up to 85%, with Norway, Denmark, and Italy leading the reductions [
8]. The success of biosimilars in Europe was due to them achieving widespread acceptance by payers, providers, and patients as an integral part of medicine through an extensive program involving preparing stakeholders, investing in evidence generation (e.g., the NorSwitch trial), and introducing incentive models to share payer savings with hospitals. A key component of this success was forced switching, which is not possible on legal grounds in some countries and due to commercial interest in others. For the most popular products, such as adalimumab, erythropoietin, filgrastim, infliximab, rituximab, etanercept, and trastuzumab, almost 100% of the market is held by biosimilars [
8].
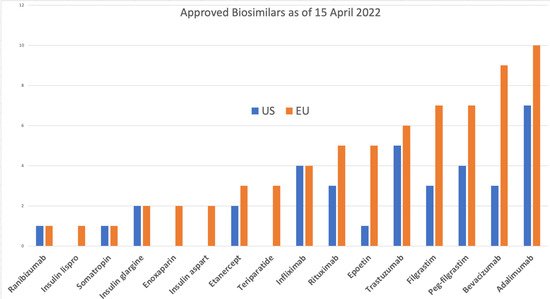
The EU has approved 84 products [
9], 70 of which are on the market, compared to 34 approvals in the US [
10], of which 19 are on the market [
11]. The reference products for all of these are only 9 molecules in the US and 17 in the EU; the exceptions in the US include insulin lispro, insulin aspart, follitropin alfa, epoetin zeta, teriparatide, and enoxaparin sodium. Enoxaparin is considered a drug by the FDA and is not reported as a biosimilar [
12]; it was approved as a drug in the EU, but now both the EU and Canada consider it a biological product that has qualified to be approved as a biosimilar. Although beginning in 2020, the FDA moved to regulate many products that were treated as drugs by the FDA, including insulins and hormones, as biologics, teriparatide is not on the FDA’s list of products that will undergo this regulatory transition [
13].
In their September 2020 report, the IQVIA Institute for Human Data Science estimated biosimilar sales to total USD 80 billion over the next five years compared to the USD 14 billion made during the previous five years (2015–2019); they also projected that the availability and use of biosimilar medicines would reduce US drug costs by USD 100 billion by 2024. In their January 2022 report, the IQVIA updated its global estimates, showing projected biosimilar sales of about USD 40 billion in 2025 and USD 75 billion in 2030 [
14].
Biologics represent 34% of pharmaceutical spending in Europe, reaching EUR 78.6 billion in 2021 and growing at a 10.5% compound annual growth rate (CAGR) over the past five years. The total European biosimilar market has reached EUR 8.8 billion in 2021 [
5], accessing between 10–40% of the total biologics market by country. By 2020, the list price savings (excluding confidential rebates and discounts) accounted for EUR 5.7 billion versus the pre-biosimilar cost of the originator. This figure would likely be even higher based on net prices.
At present, there are substantial commercial opportunities for biosimilar companies. However, with the anticipated lowering of the cost in the market, the level of competition is expected to rise, which will eventually benefit patients. Price reductions of 70% to 80% are needed to encourage the adoption of biosimilars globally.
2. Target Molecules
With 12 molecules approved as biosimilars in the US and 19 (including variants of erythropoietin) in the EU, there remains a large portfolio of potential biosimilars that are out of patent, some for a very long time (
Figure 1). Moreover, many more are going out of patent soon [
15], opening the door for many more to enter the market (
Table 1). Over the next 10 years, most biologic patent expiries will affect oncology biologics (29%), followed by biologics used to treat blood and lymphatic conditions (21%).
Table 1. Potential biosimilar candidates with expired or near expiring patents *.
| Biological Products |
| Abatacept |
Abciximab |
Aflibercept |
Alemtuzumab |
| Alirocumab |
Atezolizumab |
Avelumab |
Basiliximab |
| Bedinvetman (V) |
Belimumab |
Benralizumab |
Bevacizumab |
| Bezlotoxumab |
Blinatumomab |
Blood factors |
Brentuximab vedotin |
| Brodalumab |
Brolucizumab |
Burosumab |
Canakinumab |
| Caplacizumab |
Cemiplimab |
Certolizumab pegol |
Cetuximab |
| Crizanlizumab |
Daclizumab |
Daratumumab |
Darbepoetin alfa |
| Denosumab |
Dinutuximab |
Dupilumab |
Durvalumab |
| Eculizumab |
Elotuzumab |
Emapalumab |
Emicizumab |
| Erenumab |
Etanercept |
Evolocumab |
Follitropin alfa |
| Fremanezumab |
Frunevetmab (V) |
Galcanezumab |
Gemtuzumab ozogamicin |
| Golimumab |
Guselkumab |
Ibalizumab |
Idarucizumab |
| Inotuzumab ozogamicin |
Insulin detemir |
Insulin lispro |
Interferons |
| Ipilimumab |
Isatuximab |
Ixekizumab |
Lanadelumab |
| Lokivetab (V) |
Mepolizumab |
ogamulizumab |
Moxetumomab pasudodox |
| Muromonab-CD3 |
Natalizumab |
Necitumumab |
Nivolumab |
| Obiltoxaximab |
Obinutuzumab |
Ocrelizumab |
Ofatumumab |
| Olaratumab |
Omalizumab |
Palivizumab |
Panitumumab |
| Pembrolizumab |
Pertuzumab |
Polatuzumab vedotin |
Ramucirumab |
| Ranibizumab |
Ravulizumab |
Raxibacumab |
Reslizumab |
| Rilonacept |
Risankizumab |
Romosozumab |
Sacituzumab govitecan-hziy |
| Sarilumab |
Secukinumab |
Selumetinib |
Siltuximab |
| Teprotumumab-trbw |
Tildrakizumab |
Tocilizumab |
Urofollitropin |
| Ustekinumab |
Vedolizumab |
|
|
This market mainly drives the current focus on a few molecules (
Figure 1; for example, Humira, with over USD 18 billion per year, has the most number of approved and pending biosimilars. However, most biological products have a market value ranging from a few hundred million to billions of dollars [
16]. The biologics market was valued at approximately USD 302.63 billion in 2020, and this figure is expected to reach USD 509.23 billion by 2026, registering a CAGR of 9.06% during the forecast period 2021–2026 [
16]. Therefore, each of the products listed in
Table 1 should be a good candidate, including the veterinary products (marked with a V) and antibody–drug conjugates that have not yet received attention from developers.
3. Patent Litigation
The patent litigation of biosimilars remains a major issue, particularly in the US, though it has a declining trend [
17]. Since the enactment of the BPCIA in 2010, 50 BPCIA cases have been filed in district courts. Many of these cases involve the same parties and biosimilar products, so they do not reflect outstanding disputes. Thus far, 29 cases have been resolved, and two are pending by Amgen (16 cases), and Genentech (15 cases) are the most active plaintiffs, representing the plaintiff side in more than half of all BPCIA litigations. Amgen is also the most prevalent BPCIA defendant (with eight cases), followed by Celltrion (with seven cases) and Sandoz (with six cases). In addition, AbbVie has been involved in BPCIA litigation (as a plaintiff in five cases and declaratory judgment defendant in one case). In 2019, BPCIA district court filings began to decrease, and this trend continued in 2020 and 2021. Whereas 2018 had a record 12 BPCIA district court filings, 2019 had 5, 2020 had 4, and 2021 had only 3 [
18]. In 2021, the FDA also sought help from the US patent office in a letter to reduce the misuse of patents, and the Patent Trial and Appeal Board (PTAB) is addressing patents listed in the Orange and Purple Books [
19]. As a result, seven adalimumab biosimilars approved in the US are blocked from entering the market until 2023 [
20].
Developers in the US can choose to engage in the patent dance, in which case the originator can pick out no more than two patents to litigate or let go, leaving the originator to sue as many times as they wish. The first choice is preferred for products already on the market as biosimilars. Older products will likely have a lower bar.
4. Biosimilar Adoption
It is now widely accepted that the current regulations and practices need significant changes to make biosimilars more accessible.
4.1. The US Scene
As of 15 April 2022, there were 35 FDA-approved biosimilars in the U.S., 21 commercially available. Industry analysts say biosimilars will reduce U.S. drug expenditure by USD 133 billion by 2025 [
21].
Ten of the thirty-three products have delayed launches primarily due to patent litigation between the reference biologic and biosimilar companies. Of the 21 biosimilars on the market, 17 are used for treatments associated with cancers, 3 are used to treat autoimmune conditions, and 1 is used to treat diabetes.
In 2021, the US government, including the FDA, continued to focus on decreasing anti-competitive behavior in the biosimilar space, lowering biologic drug prices, and providing further guidance to clarify the regulatory pathway for biosimilars. Examples of the actions taken are listed below.
-
“Anti-competitive practices, such as making false or misleading statements comparing biological reference products and biosimilars, may be slowing progress and hampering the uptake of these important therapies”, quoted from an FDA and Federal Trade Commission (FTC) joint statement [
22] made in February 2020.
-
The FDA also agreed to “take appropriate steps to address companies making false or misleading communications about biologics, including biosimilars and interchangeable products, which will help deter anti-competitive behavior in the biologics market and lead to the use of all available biological products”, according to the statement. In a news release dated 20 July 2021, the FDA stated that Amgen is making false claims regarding its Neulasta medicine being more effective in its new delivery system Onpro, citing this joint statement [
23].
-
President Biden signed an executive order titled “Promoting Competition in the American Economy” [
24], which directs the Federal Trade Commission to issue rules to prevent “unfair anticompetitive conduct or agreements in the prescription drug industries, such as agreements to delay the market entry of generic drugs or biosimilars”. The order also directs the FDA to address several issues affecting biosimilars, including: (1) “improving and clarifying interchangeability standards for biological products”; (2) “supporting biosimilar product adoption by providing effective educational materials and communications to improve understanding of biosimilar and interchangeable products among healthcare providers, patients, and caregivers”; and (3) “facilitating the development
(by sponsors) and approval
(acceptance) of biosimilar and interchangeable products among healthcare providers, patients, and caregivers”. Status: enacted.
-
A new law, the “Advancing Education on Biosimilars Act” [
25], now calls for the government to provide educational materials to healthcare providers, patients, and the general public to increase awareness, knowledge, and confidence in the safety and efficacy of approved biosimilars. Status: enacted.
-
The “Star Rating for Biosimilars Act” [
26], recently presented, adds a qualification system to Medicare plans. Status: introduced.
-
The “Bolstering Innovative Options to Save Immediately on Medicines” (BIOSIM) Act [
27] intends to lower biologic drug prices by temporarily increasing reimbursement to ASP plus 8% (from ASP plus 6% previously) for providers that employ a biosimilar that is less expensive than the reference product. Status: introduced.
-
The “Preserve Access to Affordable Generics and Biosimilars Act” [
28] changes the Federal Trade Commission Act to presumptively render anticompetitive “pay-for-delay” (also known as “reverse-payment”) settlement agreements that prohibit or delay the introduction of generic pharmaceuticals or biosimilars. Status: enacted.
4.2. The European Scene
Unlike in the US, distribution, pricing, and adoption are governed by individual states in the EU individual member states, leading to large variations in biosimilar pricing and uptake [
29,
30].
Patents are not a significant issue in the EMA filing, and the litigation is left to the claiming parties. The patent laws in the EU are also different. The exclusivity for biological drugs is 10 years in the EU and 12 years in the US, giving the EU filings at least a two-year head start [
31]. However, the 10 years of exclusivity for patents and other exclusivity rights can last longer than 10 years after market approval. In the EU, process patents are rarely awarded, reducing the large barrier experienced by US filings, where the patent dance involves the product and a multitude of process patents. The differences in the patent laws between the US and the EU have a significant impact on the speed and scope of introduction of biosimilars [
32].
This entry is adapted from the peer-reviewed paper 10.3390/biologics2020009