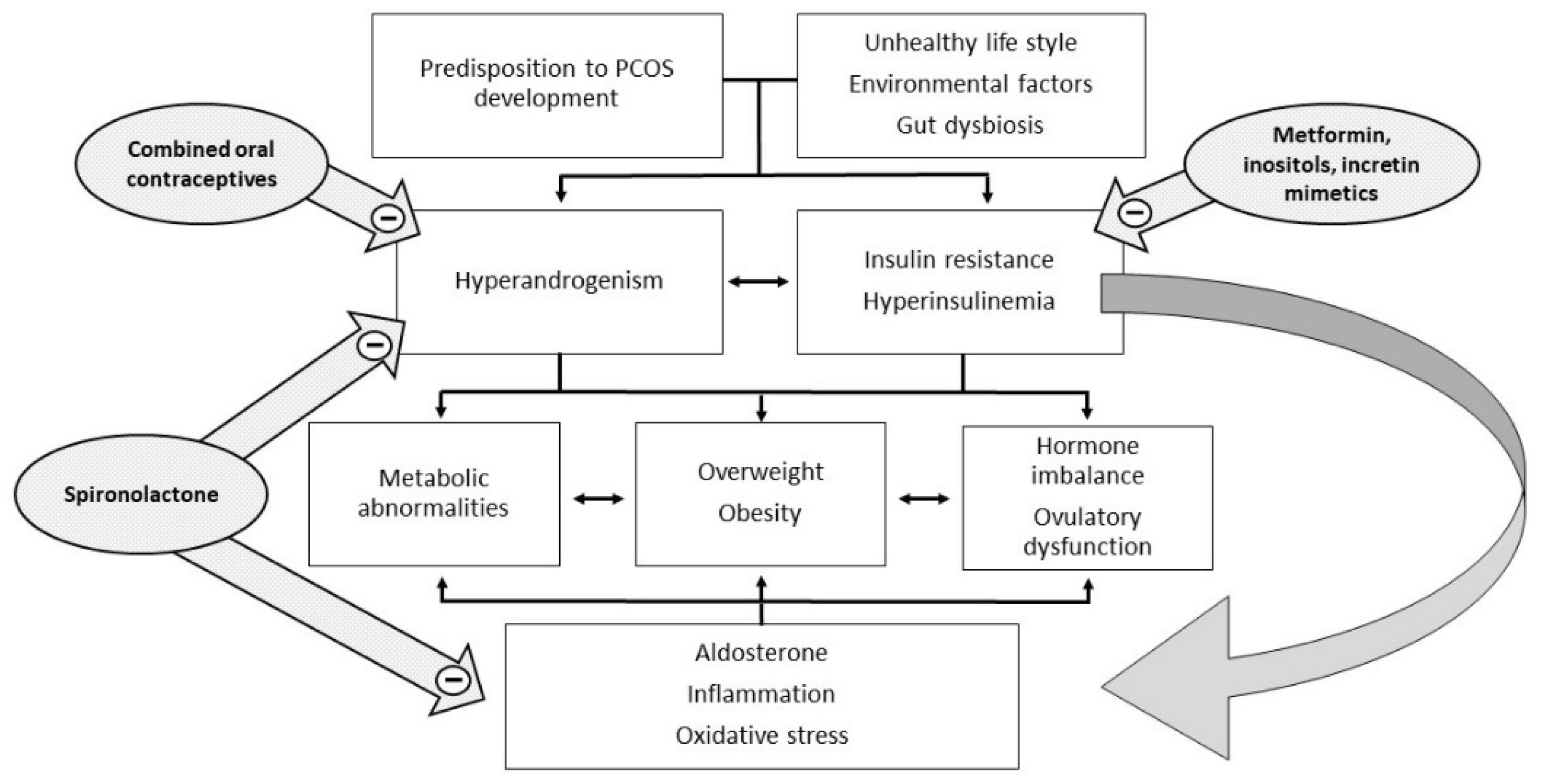
Polycystic ovary syndrome (PCOS) is the most common endocrine disease in women of reproductive age. Its prevalence varies greatly depending on the diagnostic criteria used, from 4–8% according to NIH/NICHD criteria, to approximately 18% according to the Rotterdam criteria
[1]. An international evidence-based guideline for the evaluation and management of PCOS more recently endorses and suggests the use of the Rotterdam diagnostic criteria
[2]. PCOS is a heterogeneous and extremely common disease, with symptoms that vary with the age of the patient and with therapies that must be tailored on a case-by-case basis. Its typical expression is represented by hyperandrogenism with ovarian dysfunction, chronic oligo-anovulation, and/or micropolycystic morphology of the ovary. Guidelines generally focus on the symptoms of PCOS, but remain more elusive regarding the mechanisms leading to the disease, such as insulin resistance, a very common condition in PCOS, which is worsened by hyperandrogenism-related adipose tissue accumulation
[3] and is involved in both the pathogenesis and the progression of the disease
[4]. Furthermore, apart from the use of estrogen–progestin, no drug has been approved specifically to counteract metabolic abnormalities and hyperandrogenism, so most drugs are administered off-label.
2. The Pathogenetic Role of Insulin Resistance in PCOS
Insulin acts as a regulator of glucose homeostasis by stimulating glucose uptake by insulin-sensitive tissues, such as adipose tissue, skeletal muscle, liver, and heart, but also by suppressing hepatic glucose production. Insulin is also able to suppress lipolysis, leading to a decrease in free fatty acid levels, which may mediate insulin’s action on hepatic glucose production. Insulin resistance is defined as a decreased ability of insulin to carry out these metabolic actions inherent in glucose uptake and production and lipolysis, thus leading to compensatory high insulin levels, both at baseline and after glucose loading, if pancreatic function is normal. There is still no consensus on the exact mechanism that leads to insulin resistance in PCOS, regardless of body mass index (BMI). An old study argued that in PCOS, the mechanism underlying insulin resistance decreased autophosphorylation of the insulin receptor following insulin binding
[5].
The mechanisms by which insulin resistance exerts its effects have only recently been well described
[6]. At a liver and skeletal muscle level, insulin resistance increases lipolysis with the accumulation of non-esterified fatty acids. The accumulation of intrahepatic lipids activates the diacylglycerol/protein kinase C axis and inhibits the insulin receptor, also affecting insulin signaling and subsequent gluconeogenesis. In skeletal muscle, the inhibition of phosphoinositide-3 kinase and phosphorylation of insulin receptor substrate 1 leads to impaired insulin signaling by altering the GLUT-4 expression and glucose uptake
[6][7]. The consequence of hyperinsulinemia compensatory to insulin resistance is an overstimulation of non-insulin-sensitive tissues, such as the ovaries. In particular, insulin and LH act synergistically on the theca cells, stimulating ovarian androgen production
[4]. In addition, insulin acts both directly as a co-gonadotropin, enhancing LH activity by stimulating the expression of receptors for LH, insulin, and IGF on granulosa cells, and indirectly by impairing the regulation of the hypothalamic–pituitary–ovarian axis. Hyperinsulinemia increases the adrenal steroid response to ACTH stimuli and decreases the synthesis of sex hormone binding globulin (SHBG) in the liver, with a consequent increase of both total and free androgen levels (
Figure 1).
Figure 1. Pathophysiology and potential therapeutic targets of PCOS.
Recent studies have hypothesized the role of the gut microbiome as a cause or effect of BMI, insulin resistance, and inflammation in PCOS. Gut dysbiosis due to poor-quality diet could cause the passage of lipopolysaccharides produced by Gram negative micro-organisms into the circulation. The consequence could be the activation of the immune system, insulin resistance, and hyperandrogenism
[8]. A recent revision of 31 studies published in the last 10 years reported reduced alpha diversity and dysbiosis in women with PCOS
[9]. Treatment of PCOS with prebiotics, probiotics, and synbiotics could have some beneficial effects on metabolic and biochemical profiles. Further studies should investigate the role of the microbiome in the pathogenesis and management of PCOS.
3. The Pathogenetic Role of Inflammation in PCOS
PCOS has also been associated with chronic low-grade inflammation, characterized by increased white blood cell count, high levels of C-reactive protein (CRP), interleukin 6 (IL-6), interleukin 18 (IL-18), monocyte chemoattractant protein-1, and macrophage inflammatory protein-1. Insulin resistance is related to inflammation. For example, an exaggerated production of tumor necrosis factor (TNF-α) produced by monocytes as a response to hyperglycemia could exacerbate the metabolic and hormonal abnormalities of PCOS
[10]. Recently, advanced glycosylation end products (AGEs) and their receptors implicated in the inflammation and oxidative stress cascades have also been found to be overexpressed in PCOS women
[11]. The release of inflammatory markers is associated with long-term metabolic complications and high cardiovascular risk
[12].
An underestimated factor in the diagnosis and treatment of PCOS is aldosterone
[13]. It has been shown that aldosterone and, in particular, the aldosterone/renin ratio are often increased in PCOS, and this accentuates the underlying inflammatory state and might be involved in the development of some metabolic and cardiovascular disorders
[14]. The researchers characterized aldosterone receptors in human mononuclear leukocytes
[15], and subsequent studies confirmed that the incubation of lymphocytes with excess aldosterone increased the protein expression of PAI 1 and p22phox, two markers of inflammation
[16]. Many studies have reported the role of mineralocorticoid receptor blockers, such as spironolactone, not only in the treatment of hyperaldosteronism and resistant hypertension, but also in the prevention of metabolic and cardiovascular complications and cerebrovascular accidents in patients with normal values of aldosterone
[17].
Another important pro-inflammatory agent involved in the pathogenesis of PCOS is adipose tissue
[18]. It is known that adipose tissue-resident macrophages release TNF-
α and IL-6, which are implicated in the induction of insulin resistance
[19]. Hyperandrogenism leads to aberrant adipose tissue functions in PCOS
[20]. Insulin resistance, hyperandrogenism, chronic low-grade inflammation, and adipose tissue hypertrophy and dysfunction may act together in a vicious cycle in the pathophysiology of PCOS. These observations need confirmation in larger studies directly assessing the presence of inflammation in the fat tissues of PCOS women
[21].
4. The Pathogenetic Role of Hyperandrogenism in PCOS
Hyperandrogenism in PCOS could be caused by defective intrinsic steroidogenesis in ovarian theca cells
[11] or by elevated LH levels due to altered regulation of the hypothalamic–pituitary axis, also influenced by insulin. Alterations in adrenal steroidogenesis due to CYP17α1 hyperactivation
[22] could also contribute to the hyperandrogenism of PCOS
[23]. Increased peripheral cortisol metabolism has also been proposed as a contributor to adrenal hyperandrogenism. Reduced cortisol levels cause inadequate negative feedback on the hypothalamic–pituitary–adrenal axis with increased pituitary ACTH synthesis and stimulation of adrenal steroidogenesis
[24]. If insulin resistance leads to hyperandrogenism and anovulation, hyperandrogenism is also recognized as one of the possible causes of insulin resistance in PCOS (
Figure 1). Androgen excess during intrauterine life or in the immediate post-natal period has been shown to accentuate visceral adiposity and insulin resistance. Hyperandrogenic PCOS phenotypes show an increased level of insulin resistance and metabolic complications
[25]. The administration of drugs with anti-androgenic activity improves insulin resistance
[26]. At the level of adipose tissue, testosterone acts by decreasing protein kinase C (PKC) phosphorylation
[27], whereas on skeletal muscle, it acts by increasing the phosphorylation of the mammalian target of rapamycin (mTOR) and ribosomal kinase S6 (S6K), leading to increased serine phosphorylation of IRS-1
[28]. These two androgen-mediated mechanisms exacerbate insulin resistance in adipose tissue and skeletal muscle, respectively.