Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Similar to genes, microRNA are also subjected to dysregulation, therefore various mechanisms need to be considered when studying miRNA dysregulation. Amongst them are the amplification of loci which leads to the overexpression of amplified genes, mutation leading to deregulated or loss of function, epigenetic control contributing to gene activation or suppression and the effects of trans activating elements on gene expressional control.
- microRNA
- miRNASponges
- antagomir
- miRNA mimic
1. Amplification of Loci
MicroRNA loci amplification is a common occurrence in various cancers (Figure 1). Amplification of the genomic region often causes overexpression of the miRNAs thus resulting in the depletion of its target mRNA [1]. This in turn leads to dysregulation in critical signaling pathways which contribute to cancer development and progression.
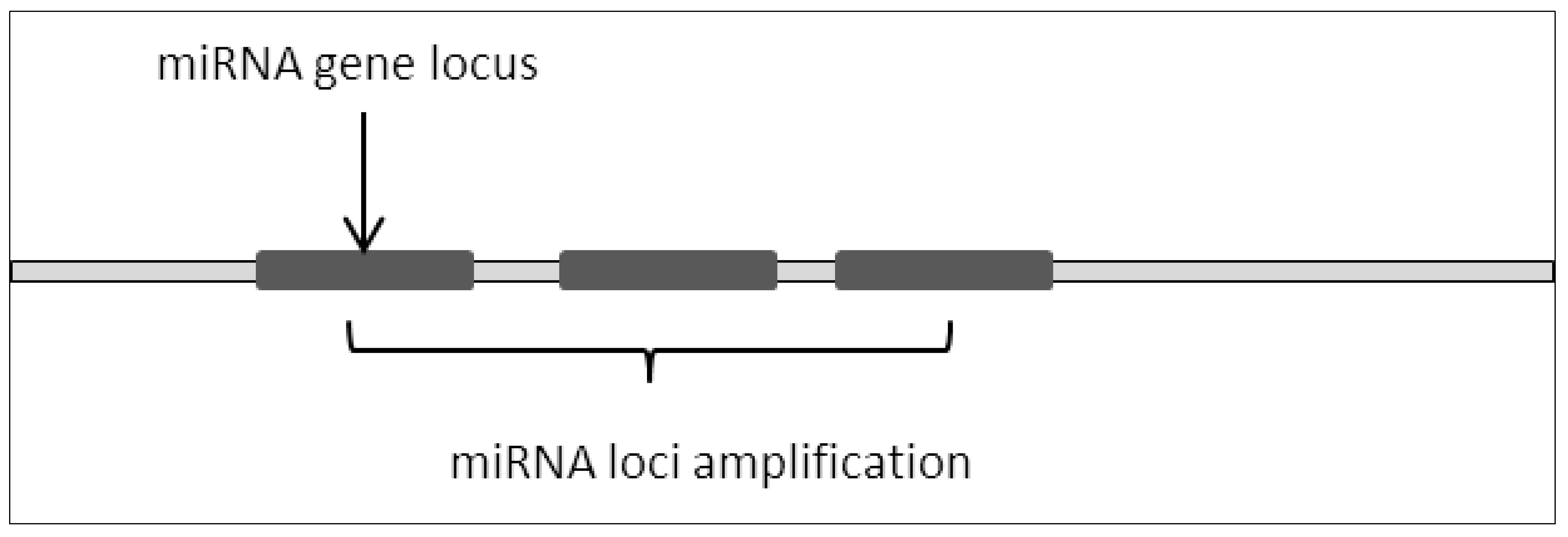
Figure 1. miRNA loci amplification resulted in repeating miRNA sequences thus increasing basal expression of miRNA.
This can be observed in embryonic brain tumors where the C19 miRNA cluster was amplified [2][3]. Interestingly, the same research also showed that the C19 cluster was fused to the TTYH1 gene, which further drives the expression of the amplified C19 cluster polycistronically. This causes extreme overexpression of DNM3TB which is an effector of the C19 miRNA cluster target, RBL2. This study clearly shows that microRNA loci amplification leads to the dysregulation of target genes and their effectors.
Amplification also occurred in the miR-17-92 cluster where it was found that the miRNAs within the cluster were significantly overexpressed in human lung cancer [4]. MiR-17-92 cluster copy number was significantly higher when compared to the copy number of miR-106a-92 and miR 106b-25. Additionally, the same cluster was also found to be amplified in acute myeloid leukemia with MLL rearrangement where it was found that this cluster affects cellular differentiation and proliferation [5].
Furthermore, miR-23a locus was also found to be implicated in the same manner in gastric cancer. Research found that the miRNA was over-expressed and that the miRNA gene was amplified along with other miRNAs (miR-1274a, miR-196b, miR-4298, miR-181c, miR-181d, miR-23a, miR-27a and miR-24-2) at loci 19p13.13. The upregulation of these miRNAs contributed to gastric cancer progression. Additionally, the group also revealed that miR-23a downregulated MT2A and that inhibition of the microRNA halted tumor growth [6].
It was also reported that amplification of miR-191/425 promotes the growth of breast cancer [7]. The group reported that the copy number of miR-191/425 was significantly higher in primary breast tumor in comparison to normal tissue. MicroRNA-191/425 target DICER1 which leads to the global miRNA dysregulation due to its function in processing pre-miRNA into mature miRNA, thus effectively leading to the promotion of breast cancer tumorigenesis.
2. Mutation (Single Point Mutation, Deletion, Insertion, Base Substitution)
Mutation to genes such as substitution or deletion often leads to the production of nonfunctional translated proteins due to impaired codon arrangements [8][9][10]. As a consequence, critical cellular signaling pathways are frequently dysregulated due to the impairment in protein structure and function where this culminates with the development of various cancers [11][12][13][14]. A similar phenomenon can be observed in miRNA where a mutation in the miRNA locus would lead to dysfunctional miRNA expression (Figure 2).
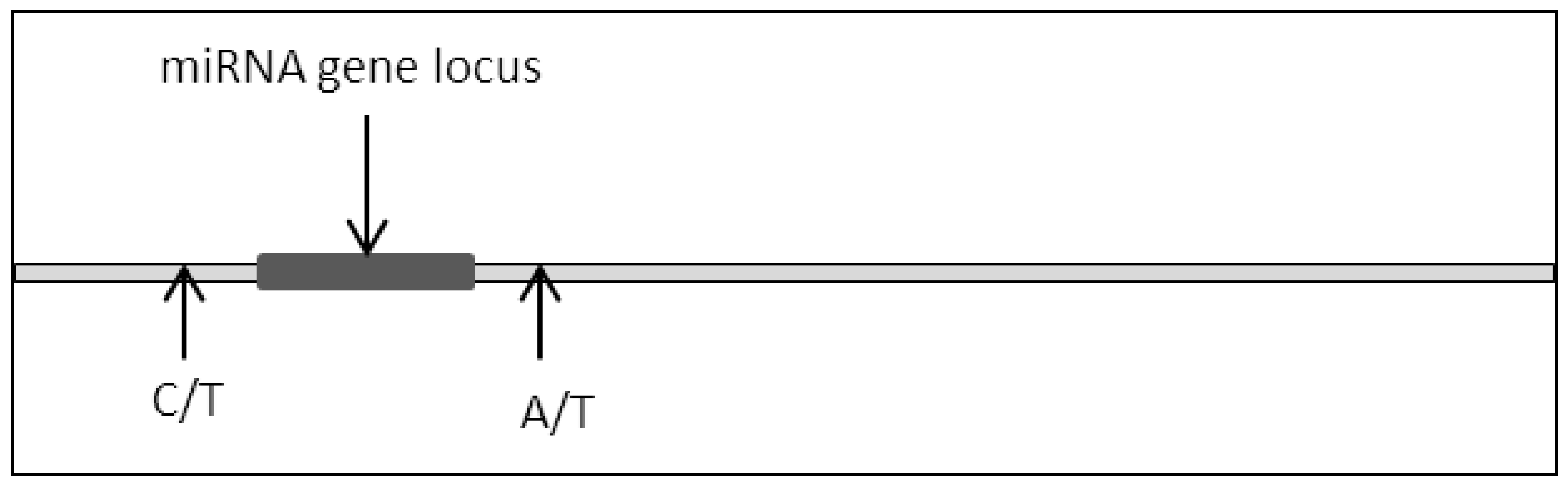
Figure 2. Single base disposition at miRNA locus.
This was proven in the case of the miR-15a and miR-16-1 locus where partial homozygous deletion of the locus led to the depletion of miR-15a expression. Furthermore, the group also reported a single base substitution of T to C 21bp upstream of the miR-15a coding sequence. It was postulated from this research that these mutations lead to the development of prostate cancer due to the prevalence of these mutations in xenografts [15].
Furthermore, point mutation of miRNA was also found to play a hand in leukemogenesis. It was found that point mutation in the miR-142 resulted in the loss of function of that miRNA. This potentiates AML towards myeloid lineages while suppressing lymphoid lineages differentiation. The loss of miR-142 leads to the accumulation of ASHL1 resulting in the maintenance of the HOXA genes. This caused the myeloid progenitors to be locked in the myeloid stage thus preventing proper cellular differentiation [16]. Additionally, it was also reported from cancer cohort (TCGA) data that the miR-142-3p seed region harbors the mutation at the 7th and 6th nucleotide where A > G and C > G/T occurs predominantly in diffuse large b-cell lymphoma [17]. Earlier results reported also that mutation in the miR-142-3p seed sequence shifted miR-142 targeting from RAC1 towards ZEB2 in diffuse large b-cell lymphoma which could attribute to disease progression [18].
3. Epigenetic Modifications on miRNA Loci
Epigenetic modulation plays a critical role in the regulation of miRNA expression [19][20]. Epigenetic markers such as DNA methylation, histone state, an abundance of CpG sites and binding of epigenetic modulators influence the accessibility of miRNA loci to various transcription machinery (Figure 3A,B). In this case, a group reported that PPARγ binds consensus sequence located on the miR-122 promoter to promote expression by inducing histone acetylation. They reported that induction of hepatocellular carcinoma cells with 5′aza-2′deoxycytidine (a compound that inhibits methyltransferase) prevented the N-cor/SMRT/HMeT complex, which is a miR-122 inhibitor from binding onto the consensus promoter sequence which in turn caused a reduction in H3K9 demethylation (heterochromatin mark on the genome, usually found to be abundant in repressed genes) and provided an accessible miR-122 locus which allowed PPARγ to bind predominantly [21].
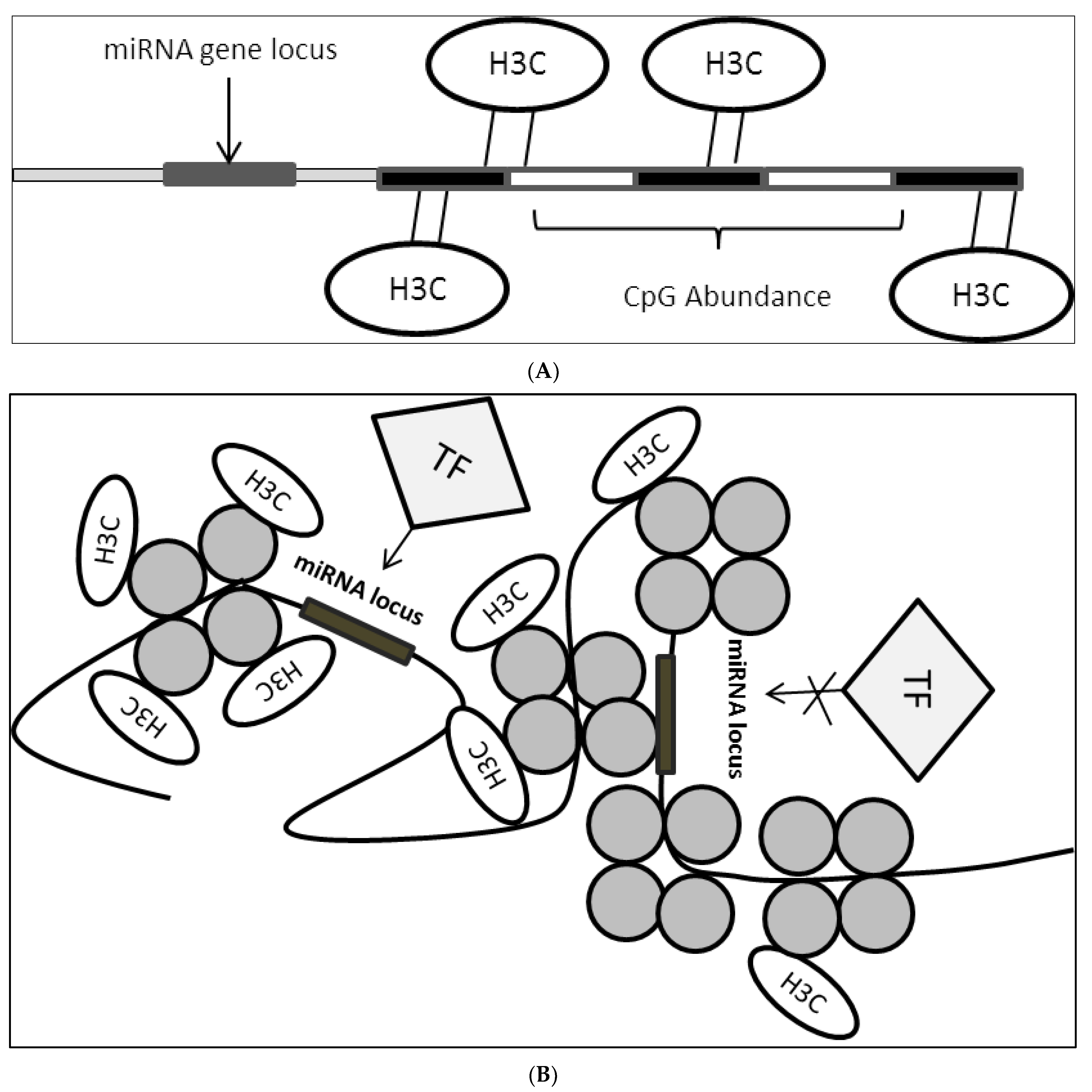
Figure 3. (A): CpG site enrichment on miRNA locus resulting in miRNA suppression. CpG methylation brought about by DNMTs methylates DNA sequence in proximity to miRNA locus or direct locus methylation leads to suppression of miRNA expression. H3C: methyl group; (B): Histone state on miRNA locus controls accessibility of TFs. Histone modification leads to the formation of open or closed chromatin structure which in turn affect TFs binding onto miRNA locus where this would result in either miRNA activation or suppression depending on the transcriptional activity of the bounded factors. H3C: methyl group.
Histone state plays a major role in the accessibility of crucial transcription machinery onto gene locus which triggers transcription or suppression. Depending on the state of chromatin brought about by the actions of histone modifiers genes can either be activated or inactivated. In miRNA regulation, it was reported that the interplay between miRNA and histone modifiers exists in modulating disease progression [22][23][24]. It was found that miR-125a-5p expression negatively regulates HDAC5 in breast cancer which contributes to disease progression via a feedback loop. The study found that miR-125a-5p binds the 3′-UTR of HDAC5 which leads to the reduction in HDAC5 dependent RUNX3 acetylation on p300/RUNX3 complex resulting in the dissociation of the complex binding on the miR-125a-5p locus. Using HDAC inhibitor, HDAC5 activity was suppressed leading to the reduction of miR-125a-5p which leads to apoptosis suggesting interplay between miRNA and HDACs in mediating apoptotic evasion [25]. In addition, it was also reported that the EZH2 histone modifier, which induces heterochromatinization, was overexpressed in pancreatic duct adenocarcinoma (PDAC) leading to the suppression of miR-218. Direct EZH2 siRNA targeting leads to restoration in miR-218 expression as this abrogates EZH2 binding onto the miRNA locus also blocking the recruitment of DNMTs via EZH2 [26].
Additionally, it was also shown that CpG sites, influence the expression of miR-335 located within the MEST gene locus downstream of MEST exon 1 which may contribute to hepatocarcinogenesis. In this study, it was found that the CpG-rich site located upstream of the miR-335 beyond exon 1 of MEST resulted in the repression of miR-335 expression due to DNA hypermethylation. The group also reveals that treatment with 5′aza-2′deoxycytidine, a known DNA demethylator, resulted in the upregulation of the suppressed miR-335 [27].
It was found that in prostate cancer overexpression of the MED1 gene would lead to the disease progression [28][29]. This effect could be mitigated via MED1 suppression by miR-205. However, it was found that the miR-205 locus was hypermethylated in prostate cancer thus leading to the epigenetic silencing of the miRNA. Additionally, the same group found that H3K9Ac, a histone mark for euchromatin (highly accessible areas within the genome usually susceptible to transcription factor binding), was significantly reduced on miR-205 loci. Furthermore, they also reported the prevalence of epigenetic in various cancer cells and found that miR-205 expression was inversely correlated with DNA methylation [30][31].
Results have shown that miR-195 was epigenetically influenced as well. It was found that the expression of miR-195, a tumor suppressor miRNA that is required for normal colon function, was downregulated in colorectal cancer cells [32][33]. It was also shown in the study that CpG islands upstream of the miR-195 locus were partially methylated in normal colorectal cells. However, in colorectal cancer, this site was fully methylated [34]. The findings from these studies suggest that DNA hypermethylation of miRNA loci in colorectal cancer plays a crucial role in oncogenesis by interfering with the tumor-suppressive activity of miR-195.
Reports have also shown that miR-340 is implicated epigenetically in neuroblastoma [35][36]. CpG-rich sites were found downstream of the predicted transcription start site (TSS) of miR-340 and these sites were extensively methylated. Interestingly, these sites were also found to overlap with RNA polymerase II (pol II) binding sites thus suggesting epigenetic modulation in transcriptional control of miRNA [37]. Treatment with 5′aza-2′deoxycytidine reduced methylation of the CpG sites which in turn led to an increase in miR-340 activity via reduction of its target gene SOX2. These findings clearly highlight the role of epigenetic modifications in miRNA dysregulation.
4. Transcription Factors (TF) Controlling miRNA Expression Dysregulation
Transcription factors (TFs) are well known to either promote or suppress gene expression [38]. Transcription factors are generally understood as proteins that bind specific consensus sequences located on genomic regions, particularly, on gene loci [39][40]. The binding of TFs can either occur proximal or distal from the TSS [41][42]. TFs that promote accessibility on the genomic region for RNA pol II binding and other TFs are known as activators whereas suppressors are TFs that promote inaccessibility [43]. The mechanism of action of TFs involves the recruitment of other TFs or epigenetic modulators on specific genomic loci. Depending on what type of TF bounded (Suppressors or Activators) they partner with different epigenome modulators (HDAC, HMET, HAT, p300). This leads to either upregulation or downregulation of genes affected [44][45].
Proper TF function is crucial for the regulation of miRNA expression. This can clearly be seen in the interaction between the transcription factor ETS5 and miR-155. It was shown that ETS5 binds to miR-155 TSS and activates the expression of the pri-miR-155 which in turn leads to lipopolysaccharide activation in the human embryonic kidney. Contrarily, the expression of miR-155 was downregulated by the addition of IL-10, an inhibitor of ETS5. This is an example of an activator mechanism in miRNA expression [46].
Additionally, it was also found that GATA3 modulates the activity of miR-29b by binding on the promoter of miR-29b [47]. Reporter assay also showed induction in expression when miR-29b loci containing the GATA3 binding sequences were cloned into luciferase plasmids [48]. However, deletion of the promoter binding sequence resulted in the reduction in luciferase activity clearly showing that GATA3 binding is crucial for miR-29b induction.
Interestingly, it was also reported that miR-1 and miR-206 were both regulated by NRF2 transcription factors in an HDAC4-dependent manner [49][50]. In this study, downregulation of NRF2 resulted in an increase in miR-1 and miR-206 expression. This indicates that the TF acts as a suppressor. NRF induces HDAC activity resulting in a reduction in accessibility (heterochromatin) on the genomic loci, thus inactivating transcription. Furthermore, downregulation of HDAC resulted in an increase in miR-1 and miR-206 expression.
A summary of the various mechanisms involved in miRNA dysregulation is presented in Table 1.
Table 1. Table showing various mechanisms of dysregulation of miRNA in cancers.
| miRNA | Mechanism of Dysregulation | Consequence | References |
|---|---|---|---|
| miR-650 | Loci amplification | Inverse correlation was observed between miR-650 and tumour supressor genes ING4 and NDRG2 | [77] |
| miR-21 | Loci amplification | miR-21 overexpression leads to PTEN suppression | [78] |
| miR-4288 | Deletion | Loss of miRNA in prostate cancer, miRNA directly represses metastatic/invasion genes MMP16 and ROCK1 | [79] |
| miR-3613 | Deletion | miR-3613 was found to be lower in breast cancer. Gain of function reveals miR-3613 to regulate PAFAH1B2 and PDK3 blocking oncogenesis | [80] |
| miR-379 | Base substitution | Hotspot mutation commonly occurring in lung adenocarcinoma | [81] |
| miR-142-3p | Epigenetic suppression via DNMT recruitment | Hypermethylation of miR-142 leads to unfavorable prognosis in nasopharyngeal carcinoma | [82] |
| miR-338-5p/421 | EZH2 mediated suppression via DNA methylation | Presence of CpG marks on primary prostate cancer. Ectopic expression reveals suppression in prostate cancer growth | [83] |
| miR-17-92/106b-25 | CMYC driven | CMYC drive the expression of these miRNA clusters, inhibition of cMYC activators resulted in suppression of these clusters in hepatocellular carcinoma | [84] |
| miR-122 | CMYC driven | CMYC oncogene overexpression in hepatocellular carcinoma activates miR-122 via direct promoter binding driving oncogenesis | [85] |
| miR-455-3p | Reside in host gene driven by p53 | miRNA involves in cancer quiescence via p53 mediation | [86] |
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10040915
This entry is offline, you can click here to edit this entry!