1. Cancer
Cancer cells attract neutrophils (tumor-associated neutrophils, TANs), which result in the release of NETs into the tumor microenvironment. NETs have been found in several types of mammalian tumors, including pancreatic, breast, liver, lung, and gastric cancers; their presence in solid tumors is associated with poorer prognosis
[1][2][3]. While NETs can exert anti-tumor actions via MPO
[4], proteinases
[1][5], and histones
[6], the proteinases can also digest the extracellular matrix (ECM), enhancing metastasis
[1][5]. Additionally, it has been shown that NETs are capable of capturing circulating tumor cells via the upregulation of β1-integrin expression, an interaction that was abrogated using DNase
[7]. Therefore, it is unsurprising that NETs have been found around metastatic tumors
[8]. In fact, citrullinated histones from NETs were found to promote cancer metastasis by binding to coiled-coil domain-containing protein 25 (CCDC25)
[9]. CCDC25 knockout cells were found to lose their adhesive properties to NET-DNA; NET-DNA-induced cytoskeleton remodeling was inhibited; and chemotaxis toward NET-DNA was reduced
[9]. Injection of the CCDC25 KO cells into immunocompromised mice significantly inhibited NET-mediated lung metastases. Contrarily, overexpression of CCDC25 enhanced cancer cell adhesion and migration toward NET-DNA in vitro, and promoted the formation of lung and liver metastases in vivo
[9]. Additionally, the authors found high CCDC25 expression in primary tumors to be associated with poor prognosis
[9]. Another group showed the ability of NETs to promote gastric cancer metastasis via the induction of epithelial–mesenchymal transition (EMT), enabling intravasation of tumor cells into the circulation or lymphatics, leading to tumor formation at a distant tissue site (Pastushenko, 2019). Using Western blot and immunohistochemical analysis, the authors showed decreased levels of E-cadherin, an epithelial adhesion marker, and increased levels of vimentin, a mesenchymal marker in human gastric adenocarcinoma cells treated with NETs
[10].
In addition to the pro-metastatic function of NETs, they have also been shown to promote tumor cell growth by enhancing their mitochondrial functions
[11]. Yazdani et al. showed that stressed cancer cells upregulate the expression of several chemokines and high-mobility group box 1 (HMGB1), recruiting neutrophils to the tumor microenvironment (TME), inducing NET formation. NETs and the NE present in the fibrous structure were demonstrated to promote mitochondrial homeostasis, increasing energy production, and accelerating cancer cell proliferation via TLR4 (Figure 1). The blockade of this interaction through genetic manipulation, pharmaceutical inhibition or siRNA targeting of the axis components were shown to abrogate mitochondrial biogenesis and impede tumor growth
[11].
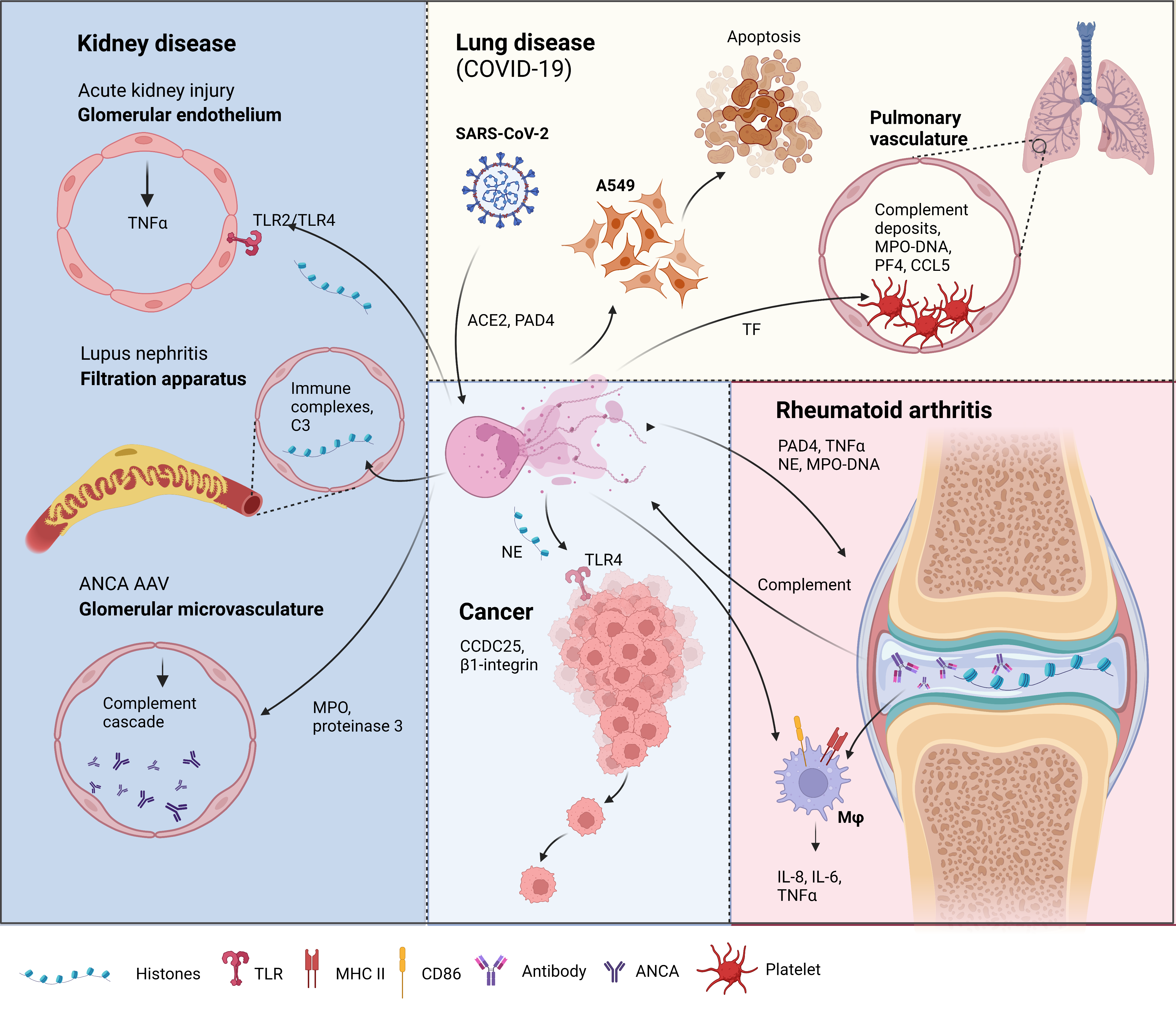
Figure 1. Neutrophil extracellular traps (NETs) in disease pathology. Acute kidney injury: NETosis in the glomerular capillaries results in histone release, triggering renal cell necrosis via toll-like receptor 2 (TLR2)/TLR4, leading to tumour necrosis factor (TNFα) release [32]. Lupus nephritis: Accumulation of immune complex deposits containing anti-double stranded DNA (dsDNA) IgG and its target, plus the complement component, C3, attract neutrophils to the renal filtration apparatus [33]. Presence of hyperacetylated histones in the immune deposits trigger an increase in reactive oxygen species independent NETosis [34]. Anti-neutrophil cytoplasmic antibodies (ANCA) associated vasculitis (AAV): Neutrophils from AAV patients undergo spontaneous NETosis [35]. ANCA against myeloperoxidase and proteinase 3, two proteins in neutrophil granules are found in the glomerular microvasculature 135 [36]. These autoantigens activate the complement cascade and induce further NETosis [37, 38]. Lung disease in coronavirus disease-19 (COVID-19): Severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) can induce NETs via an angiotensin converting enzyme 2 (ACE2) and PAD4 dependent mechanism [39]. These NETs can enhance the apoptosis of a human alveolar basal epithelial cell line, A549 [4]. Complement deposits in the pulmonary vasculature prime neutrophils for NETosis, resulting in myeloperoxidase (MPO)-DNA complexes. Elevation of platelet factor 4 (PF4), chemokine ligand 5 (CCL5) [40], and the neutrophil-platelet interaction all trigger NETosis [41, 42], which was shown to have thrombogenic activity via the upregulation of tissue factor (TF)[41]. Rheumatoid arthritis (RA): Peptidylarginine deiminase 4 (PAD4) citrullinates histones released by spontaneous NETosis, forming immune complexes in the synovial joint that activate macrophages (mφ)[43]. The immune complexes can also activate the complement cascade, which tiggers, further NETosis [44]. NETs from RA donors can activate resting macrophages to induce the surface expression of major histocompatibility complex II (MHCII) and cluster of differentiation 86 (CD86)[45]. These macrophages also release interleukin-6 (IL-6), IL-8, and TNFα [45], which can feedback to induce more NET formation. Cancer: Cancer cells attract neutrophils into the cancer microenvironment (CME), which results in NET formation under the inflammatory CME. These NETs can capture circulating tumour cells via β1-integrin [46], citrullinated histones from NETs can also enhance cancer metastasis by binding to coiled-coil domain containing protein 25 (CCDC25)[47]. NETs and the neutrophil elastase (NE) released can promote tumour cell growth via TLR4 [48].
2. Multiple Sclerosis
Myelin damage in the white matter may result from the failure to maintain compact myelin, resulting from abnormally enhanced citrullination of myelin basic protein (MBP), which form less cationic isomers, thereby destabilizing the myelin multilayers
[12]. This also enhances the susceptibility of the myelin sheath to digestion by proteases
[13]. Additionally, citrullinated MBP causes fragmentation of lipid vesicles of the myelin layer, which may contribute to further demyelination
[14][15]. Furthermore, studies have found the degree of MBP citrullination to correlate with disease severity
[16][17]. Unsurprisingly, increased levels and enzyme activity of PAD4 have been found in the central nervous system (CNS) of multiple sclerosis (MS) patients and animal models, with an associated increase in citrullinated histone observed in the white matter
[18][19]. Nuclear PAD4 and TNFα were shown to be elevated before the onset of the demyelinating disease in mouse models
[20], suggesting their value as prognostic markers for MS. Furthermore, an elevated level of TNFα was detected to be released by astrocytes and not infiltrating immune cells
[19], which may contribute further to local inflammation by priming neutrophils for NET formation.
Current clinical evidence suggests that neutrophils may contribute to MS pathogenesis, as they are not only found in increased frequency in MS patients, but the neutrophils are also in an activated state with a reduction in apoptosis. These neutrophils also have increased levels of TLR2, IL8R, enhanced degranulation, and oxidative burst
[21]. Elevated levels of NETs have been detected in the serum of MS patients
[21], with NE levels directly correlating with disease severity and clinical prognosis
[22], while significantly more MPO-DNA were complexes detected in male patients, who tend to have poorer prognosis than their female counterparts
[21][23]. Pharmaceutical intervention may also play a role in disease progression, where patients on corticosteroid treatment were found to have significantly increased NE activity, whereas patients on IFNβ-1b treatment showed a lower level of NE and immune complexes
[24].
The most recent elucidation of the mechanistic role of NETs in MS pathogenesis was performed by Wilson et al., showing that histones can directly activate T cells for T-helper 17 (Th17) differentiation via TLR2 and myeloid differentiation primary response 88 (MyD88), leading to the phosphorylation of signal transducer and activator of transcription (STAT3)
[1], essential for Th17 cell responses
[2]. Komiyama et al. also demonstrated the ability of NETs to activate inflammatory Th17 cells, which can secrete IL-17, a neutrophil chemotaxis cytokine
[25]. Using IL-17
−/− mice, the authors showed a significant decrease in immune cell infiltrate, delayed the onset and reduced disease severity
[25], providing a potential therapeutic target for MS treatment. Other groups have demonstrated that pharmaceutical inhibition of MPO reduced oxidative stress and enhanced integrity of the blood–brain barrier (BBB), decreasing disease severity
[26][27].
3. Kidney Disease
The kidney is prone to immune responses that lead to the development of renal injury and chronic kidney disease progression
[28][29][30][31]. This can trigger neutrophil infiltration to the site of injury, compounded by the damage-associated molecular patterns (DAMPs) released by the necrotic renal cells, inflammation and tissue damage become amplified in the kidney
[29][32]. The released DAMPs then go on to stimulate the infiltrating neutrophils and induce PAD4 activation, which triggers chromatin decondensation and NET formation
[33].
3.1. Acute Kidney Injury (AKI)
Acute kidney injury can occur as a consequence of necroinflammation, an interplay of injury and inflammation, where complement activation, exotoxins, or cytotoxic T cells can trigger renal cell necrosis
[32]. Kumar et al. showed that NETosis results in histone release, leading to glomerular endothelial cell death in an TLR2/TLR4-dependent manner (Figure 1)
[34]. EC histones also trigger the release of TNFα in the glomerular capillaries
[34], a cytokine that is not only an inducer of NET formation
[35], but also characterized to be prothrombotic
[36][37][38]. This study provided evidence for the presence of fibrinogen within the glomerular capillaries, which was abrogated with an anti-histone IgG
[34]. Damage by histone release from NETosis was further characterized by Nakazawa et al. to show that histone citrullination is essential in inducing NET formation
[33]. Additionally, tubular cells killed via NETosis released factors that triggered further NET formation
[33], creating an auto-amplification loop for inflammation.
3.2. Lupus Nephritis
NETosis is commonly found in active systemic lupus erythematosus (SLE)
[39]. One of the most serious complications of SLE is nephritis, inflammation of the renal filtration apparatus, with chronic autoimmune insults to the glomerulus eventually leads to kidney failure
[40][41][42]. A key characteristic of lupus nephritis is immune complex deposits containing anti-double-stranded DNA (dsDNA) IgG and its target, along with C3 accumulation, both attracting inflammatory cell infiltrates
[39]. Furthermore, the presence of dsDNA can activate type I IFN signaling via the cGAS-STING pathway
[43], enhancing the inflammatory response (Figure 1).
Presence of NETs in renal biopsies from lupus nephritis patients may indicate their contribution to kidney damage
[44]. Rother et al. revealed that hyperacetylated histones were present in the SLE immune deposits and stimulated an increase in ROS-independent NETosis within 30 min
[45]. They also showed that immune deposits from patients with active lupus nephritis displayed the highest acetylation levels and were the most potent at inducing NETs compared to SLE patients without nephritis
[45]. Furthermore, SLE patients with nephritis or prior nephritis have an inability to degrade NETs due to impaired DNase activity
[45]; thus, existing NET deposits can stimulate further inflammation. This impaired NET removal from patient serum correlates with proteinuria, decreased albumin levels, and lower creatinine clearance rates, all indicators of active nephritis
[46][47].
3.3. ANCA-Associated Vasculitis
ANCA-associated vasculitis (AAV) comprises part of a group of immune vasculitides, characterized by necrotizing inflammation of small vessels and circulating ANCAs
[48]. The three types of AAV include granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA)
[49]. The presence of ANCA against myeloperoxidase (MPO) and proteinase 3, two proteins in neutrophil granules, is characteristic of ANCA-associated vasculitis
[50]. As discussed previously, both autoantigens are present in NETs, and have been characterized to increase cell adhesion, activate the complement cascade, and induce NET production
[51][52]. MPO is recognized as an autoantigen by MPO-specific autoantibody, resulting in a vicious cycle of inflammation where the generation of circulating ANCAs activate neutrophils (Figure 1), which deposit MPO in the glomeruli and recruit macrophages and CD4 T cells, resulting in inflammation and the induction of glomerulonephritis. This manifests clinically as MPA
[53], an ANCA-associated pathology that affects the small vessels in the renal glomeruli in particular, but can also effect the blood vessels in the lungs and skin
[54]. Immunoglobins purified from MPA patients have demonstrated the ability to induce NETs in direct correlation with the affinity of ANCA for MPO
[53]. DNAse I activity is also lower in MPA patients, indicating insufficient degradation of NETs
[55], which has been found to be responsible for the renal damage observed in up to 90% of MPA patients
[56].
Although the current paradigm assumes that ANCAs induce NET formation, neutrophils from AAV patients are not prone to apoptosis, but can spontaneously undergo NETosis
[56]. Furthermore, recent data have shown that NETosis in AAV is independent of serum ANCA levels
[57]. Additionally, excessive NET formation in AAV is directly correlated with active clinical disease rather than severe infection, implicating the significant role of NETs in autoimmunity
[57].
3.4. Goodpastures Disease (Anti-Glomerular Basement Membrane Disease)
Akin to lupus nephritis and ANCA-AAV, the pathogenesis of anti-glomerular basement membrane (GBM) disease involves both immune complex deposition and microvascular damage
[58]. These immune complexes stimulate NETosis, triggering FcγR-mediated endocytosis and tyrosine-protein kinase ABL/proto-oncogene tyrosine-protein kinase Src (Abl/Src) kinase activation, leading to glomerular endothelial injury
[59][60], where disease could be attenuated by inhibiting Abl/Src signalling in a bovine model
[60]. A murine anti-GBM glomerulonephritis model showed the presence of NETs in a necrotising lesion, where histones from NETs were shown to cause capillary injury, which was ameliorated using PAD inhibitors and anti-histone antibodies
[34]. Contrarily, research by another group found that PAD4 inhibition using either antibody or genetic deletion had no protective effect in anti-GBM disease
[61], challenging the role of NETs in anti-GBM pathogenesis.
3.5. Vasculitis
Vasculitis is a broad term encompassing diseases involving inflammation of the blood vessels
[62], it includes ANCA-associated vasculitis (AAV, small-vessel vasculitis), Takayasu arteritis (large-vessel vasculitis), and giant cell arteritis
[63]. Contention exists around whether NETosis is a hallmark of active AAV as high levels of NET remnants are found in all three types of vasculitis mentioned above
[56]. However, proteinase 3-ANCA (PR3-ANCA) and MPO-ANCA have been found in most vasculitis patients, both characteristic of AAV pathogenesis
[64]. Several studies have shown that autoantibodies, serum immune complexes, and IgG derived from patients with AAV have the capacity to induce NETosis
[44][55][65][66][67][68]. Recent work has shown that MPO-ANCA may cause NETosis in patients with vasculitis due to its affinity rather than the antibody levels
[55][66][69]. These findings are further supported by a renal biopsy study, where individuals with high-affinity MPO-ANCA demonstrated enhanced levels of NETosis
[57].
PR3-ANCA and MPO-ANCA can cause NET formation in patients with AAV
[55]; however, the mechanism by which this occurs remains unclear. As discussed previously in this review, NETosis requires neutrophil activation, an event characterized by the respiratory burst, which was induced by crosslinking PR3-ANCA or MPO-ANCA to the cell surface
[70]. Furthermore, many factors released by neutrophils can activate the complement cascade
[71][72][73], and several animal studies have shown that C5a–C5aR interactions can prime neutrophils for activation by ANCA
[74][52][75]. C5aR1 may be a promising therapeutic target, as inhibition with antagonist CCX168 (Avacopan) has demonstrated efficacy in mediating disease severity
[75]. Other autoimmune studies have found that NETosis may be negatively regulated by the interaction between neutrophil semophorin D and plexin B2 on endothelial cells, this is further evidenced by the ability of recombinant plexin B2 in the blockade of NETosis in human cells.
4. Rheumatoid Arthritis
As discussed earlier in this review, NETosis is distinguished from other forms of cell death by the presence of citrullinated histones, a PAD4-mediated process. The involvement of PAD4 in the citrullination of histones was uncovered in animal models of rheumatoid arthritis (RA), where the presence of PAD4 and citrullinated proteins showed a positive correlation with disease progression
[76]. RA is characterized by the generation of autoantibodies against citrullinated proteins, which form immune complexes with citrullinated fibrinogen in the synovial tissue, resulting in the activation of macrophages (Figure 1)
[77]. As citrullinated histones are found within NETs, as mentioned earlier on in this review, neutrophils undergoing NETosis may also become a target; therefore, any areas of local inflammation can become amplified with autoantibody binding and further exacerbation of injury. This process can become a self-perpetuating cycle, where autoantibodies targeting the citrullinated histones in NETs can also activate the complement system
[78], releasing products from the complement cascade that prime neutrophils for NETosis, as discussed previously. This may provide an explanation for the accepted paradigm where complement activation at the site of autoantibody binding of citrullinated proteins can result in tissue damage
[79].
NETs from RA patients induce stronger inflammatory response compared to NETs from healthy donors
[35]. Furthermore, neutrophils from RA patients showed increased spontaneous NET formation in vitro, with potential for elevated ROS production, increased NE and MPO expression, PAD4-mediated citrullination of H3. This was all abrogated with IgG depletion, but restored with an anti-citrullinated peptide antibody
[80]. Additionally, NETs from both healthy and RA donors were shown to activate resting macrophages, inducing the upregulation of human leukocyte antigen (MHC) class I/II and cluster of differentiation 86 (CD86) on the cell surface
[35]. Furthermore, NET stimulation of the resting macrophages induced the secretion of IL-8, IL-6, and TNFα
[35], all proinflammatory cytokines that have been characterized to be dysregulated in RA and other autoimmune conditions
[81][82][83][84], where antibodies targeting the latter two cytokines are currently used in the clinic for the treatment of RA
[84][85]. Despite the pathogenic role of NETs in RA, Ribon and colleagues showed that they have anti-inflammatory effects on LPS-activated macrophages, inhibiting the release of IL-6
[35]. The authors concluded that NETs can trigger both pro- or anti-inflammatory effects depending on the target cell, its activation status, and the stimulus
[35].
5. Lung Disease
While NETs in the pathogenesis of lung disease have been extensively characterized, recent research has provided evidence for their involvement in severe acute respiratory syndrome-related coronavirus (SARS-CoV-2) infection. A higher abundance of circulating neutrophils has been correlated with a worse clinical outcome in coronavirus disease 2019 (COVID-19)
[86][87]. As NETosis is an important effector function of neutrophils under inflammatory conditions, it was not surprising that MPO and histone 3 (H3), both NET components, levels were found to be elevated in the plasma, tracheal aspirate, and lung biopsies from COVID-19 patients
[88][89]. Their neutrophils were capable of extruding more NETs, which were also larger than those produced by neutrophils from healthy controls
[88]. SARS-CoV-2 induction of NETs is dependent on angiotensin-converting enzyme 2 (ACE2), serine protease, viral replication, and PAD4 (Figure 1)
[88]. The release of NETs from the SARS-CoV-2-activated neutrophils was found to enhance the apoptosis of A549 cells, a human alveolar basal epithelial cell line (Figure 1)
[88], which can compromise lung function and lead to further disease progression.
Thrombogenic events have been documented in severe COVID-19 patients to induce lung damage
[90]. Significant deposits of complement components have been found in the pulmonary microvasculature of severe COVID-19 patients with respiratory failure
[90]. As discussed previously, the presence of complement elements can prime neutrophils for NETosis, with the addition of the cytokine storm observed in these patients
[91], another NET inducer, it may not be unreasonable to assume that NETosis is contributing to microvasculature damage. This has been evidenced by a recent study, where NETs were found to colocalized with platelets in pulmonary vasculature from COVID-19 autopsies and disease severity correlated directly with MPO-DNA complexes
[92]. Furthermore, there was a significant elevation of platelet-derived factors such as platelet factor 4 (PF4) and chemokine ligand 5 (CCL5 or RANTES) that activate NET formation
[92]. Another study characterized the interaction between COVID-19 and C5aR1, where MPO-DNA complex levels were found to correlate with thrombin/antithrombin (TAT) activity, suggesting activation of the thrombin axis
[93]. This neutrophil–platelet interaction is necessary for NET formation in thromboinflammatory disorders
[94][95], their presence was detected in aggregates in the circulation by Middleton et al.
[92]. Additionally, NETs have demonstrated thrombogenic activity in many inflammatory disorders via the expression of functionally active tissue factor (TF)
[94]. Skendros et al. found that platelet-rich plasma-stimulated neutrophils induced the expression of TF mRNA, efficiently produced NETs bearing TF, which showed high TAT activity
[93]. As C5a is key mediator of neutrophil TF expression
[96][97][98], Skendros and colleagues showed that selective blockade attenuated TF expression and functionality, inhibiting NET release
[93].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23073793