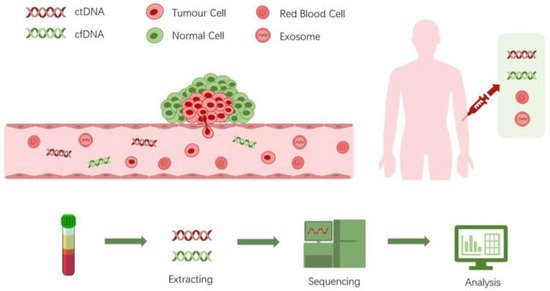
Liquid biopsy with circulating tumor DNA (ctDNA) profiling by next-generation sequencing holds great promise to revolutionize clinical oncology. It relies on the basis that ctDNA represents the real-time status of the tumor genome which contains information of genetic alterations. Compared to tissue biopsy, liquid biopsy possesses great advantages such as a less demanding procedure, minimal invasion, ease of frequent sampling, and less sampling bias. Next-generation sequencing (NGS) methods have come to a point that both the cost and performance are suitable for clinical diagnosis. Thus, profiling ctDNA by NGS technologies is becoming more and more popular since it can be applied in the whole process of cancer diagnosis and management.
1. Introduction
It is now well known that a tumor can shed cancer cells, extracellular vesicles, proteins and nucleic acids into the peripheral blood [
1]. These tumor-derived substances possess great potential as therapeutic targets or diagnostics biomarkers (
Figure 1). The idea of liquid biopsy is to sample the biomarkers found in the non-solid biological tissue for analysis of disease status. Different biological substances such as circulating tumor cells (CTCs), cell-free nucleic acids, exosomes, or proteins can be sampled and analyzed in various body fluids such as blood or urine [
2,
3]. In particular, the blood is the most widely used sampling source of liquid biopsy due to the rich information it carries and because it is easy to access [
4]. Among these blood-borne biomarkers, circulating tumor DNA (ctDNA) has gained a tremendous amount of interest in recent years. In this review, we discuss the biology, clinical utilities, and applications of ctDNA as biomarkers, emphasizing the technologies for their detection.
Figure 1. Deciphering of liquid biopsy.
2. Biology of ctDNA
ctDNA is shed into the peripheral blood by tumor cells in cancer patients. However, the biological mechanisms of how ctDNA enters and presents in the blood circulation have not yet been fully elucidated [
5]. Generally, it is widely accepted that apoptosis and necrosis of tumor cells are the main sources of ctDNA. Studies have revealed that ctDNAs are generally more fragmented than nonmutant cell-free DNA (cfDNA), with a maximum enrichment of fragments between 90 and 150 bp [
6] while the half-life of ctDNA ranged from only 15 minutes to a few hours [
7]. Although it is challenging to preserve and use ctDNA as a biomarker because of the short half-life time window, it may reflect a snap-shot of the tumor genome status, providing a way to monitor the disease. However, ctDNA represents only a small part of cfDNA in the circulation. Several biological processes are known to release cfDNA, including phagocytosis, active secretion, neutrophil release, excision repair, and pyroptosis [
8]. Thus, discriminating ctDNA from the normal cfDNA is vital to precisely reflect the disease status. Since ctDNA is derived from malignant tumor cells, they carry the tumor-specific abnormality that is not present in the normal cfDNA. The tumor-specific abnormalities that ctDNA possesses are inherited from its tumor origins, such as single-nucleotide mutations, epigenetics changes, and copy number variations [
9]. These molecular features can then be used as specific biomarkers for cancer monitoring and management references.
Apart from tumor-specific molecular characteristics, studies have also found that the levels of ctDNA in advanced stage cancers such as breast, colorectal, pancreatic, and gastroesophageal cancers are higher than in the early stage of diseases [
10]. However, the amount of ctDNA is not simply associated with the tumor size, but varies in different types of cancer [
11,
12]. Furthermore, ctDNA can both derive from primary tumor and metastases, thus is associated with the pathological status of the disease [
13]. Because of the different origins of ctDNA, liquid biopsy together with ctDNA analysis may be a better representation of tumor genomic diversity compared to biopsy from a single solid tumor sample [
14]. In other words, ctDNA reflects a better overview of the most up-to-date status of the tumor genome, making it superior to tissue biopsy analysis for cancer patient diagnosis and management. Circulating cell-free RNA (ccfRNA), mainly including mRNA and miRNA, is another type of cell-free nucleic acid that can be found in the blood stream, and also possesses potential to become a biomarker for cancer [
15]. Although ccfRNA shares many similar characteristics to ctDNA, there are more challenges when using them as cancer biomarkers [
16]. Thus, more studies and technical advances are still needed before ccfRNA can become as popular as ctDNA in cancer diagnosis. Therefore, we focus on the discussion of ctDNA in this review.
3. Next Generation Sequencing Technologies for ctDNA Detection
Because the ctDNA represents only a small part of the cfDNA, it is important to utilize the tumor-specific alterations to distinguish themselves from their wild-type counterparts. Currently, most methods using ctDNA as cancer biomarkers focus on mutation de tection [
17]. Mutations in ctDNA can be detected either by PCR or NGS [
18,
19]. However, PCR approaches only detect known mutations in certain genes. Patients without those mutations will be left out, limiting their applications as a generic diagnosis technique for ctDNA analysis. NGS approaches, on the other hand, cover a broader range of mutations by examining the whole sequences of genes of interest. In this review, we focus on discussing the NGS approaches for ctDNA profiling.
NGS for ctDNA analysis can be targeted or untargeted as well [
20]. Targeted approaches for ctDNA profiling usually sequence dozens of genes to hundreds of genes or even the whole exome. Because of relatively low throughput, high sensitivity can then be achieved by deep sequencing specific regions of interest that cover clinically relevant mutations. To enrich targets, either multiplex PCR or hybridization capture strategies are ap plied to ensure maximal amplification of target gene fragments. Due to better specificity and sensitivity, targeted sequencing is more suitable for clinical diagnosis. In the case of untargeted approaches, no enrichment step is performed, thus sequencing the whole genome. Although whole-genome sequencing scarifies the sequencing depth, it can be used to discover new genomic aberrations related to patient prognosis or treatment strategies, making it suitable for basic biomedical research.
For the sequencing step, the cost, accuracy, and speed are all key factors that need to be taken into consideration for ctDNA profiling. It is particularly true for diagnosis purposes. Under this circumstance, there are currently several platforms more suitable for ctDNA sequencing, including Illumina’s sequencing-by-synthesis (SBS), Thermo Fisher Scientific’s Ion Torrent, and the nanopore sequencing from Oxford Nanopore Technologies [
21,
22]. Illumina’s sequencing platform is dominating due to its high throughput and high accuracy. The Ion Torrent semiconductor sequencer is lower in throughput but faster in the run speed, making it advantageous when speed is required. During the past decade, nanopore sequencing has been improving in its accuracy but is still lower than the other mainstream sequencers. However, it is advantageous in the convenient library preparation and sequencing procedure. Thus, in the case when the requirement of accuracy is not high, such as profiling copy number variations, nanopore sequencing can be used for ctDNA analysis [
23]. After generation of raw sequencing data, applying the right bioinformatic algorithms is also crucial for mutation detection. A review that summarized bioinformatics tools for ctDNA analysis can be found here [
24]. There are also databases that can be used to help with the interpretation of the liquid biopsy data. liqDB is a database for liquid biopsy small RNA sequencing profiles that provide users with meaningful information to guide their small RNA liquid biopsy research and to overcome technical and conceptual problems [
25]. ctcRbase is a database that can be used for query and browse gene expression data of circulating tumor cells/microemboli [
26].
The key for precisely identifying ctDNA from a large amount of background cfDNA is to correctly detect the alterations of ctDNA compared to normal cfDNA and single nucleotide mutation is the most commonly detected alteration [
27]. Technically, in the context of ctDNA sequencing, the sensitivity of detecting the so-called mutant allele frequency (MAF) or variant allele frequency (VAF) is often used to evaluate the performance of an NGS ctDNA profiling assay [
13,
28]. MAF or VAF describes the proportion of DNA molecules containing a mutation over the total number of molecules containing the same allele. Thus, they reflect the amount of ctDNAs when referring to the cfDNAs that contain tumor-specific mutant alleles. Therefore, the lower the MAF can be detected, the more sensitive an NGS assay is for ctDNA analysis.
4. Strategies for True Mutation Detection Using NGS
Because ctDNA is mainly distinguished from normal cfDNA by tumor-specific mutations, it is thus critical to identify a true mutation for sensitive ctDNA detection. Plus, the low levels of ctDNA presented in the bloodstream also warrant highly sensitive and specific methods that can detect and quantify mutant alleles in cfDNA. This can be achieved both by experimental designs and bioinformatics algorithms. Some of these strategies are presented below to elucidate the general principles.
Tagged-amplicon deep sequencing (TAm-Seq) introduces a two-step amplification strategy to first amplify regions of interest by multiplex PCR using specifically designed primers at low concentrations to avoid inter-primer interactions, followed by using a microfluidic system in a second amplification step to amplify individual target regions using the amplicons from the first step [
29]. Sequencing adaptors were attached to the amplification products and unique molecular identifiers were used to tag different samples by another PCR step. Tam-seq was able to identify mutations of MAF as low as 2% with sensitivity and specificity over 97% [
29]. The enhanced TAm-Seq (eTAm-Seq) can detect MAF as low as 0.25% with a sensitivity of 94%. It also utilizes revised bioinformatics analysis to identify single-nucleotide variants (SNVs), short insertions/deletions (indels) and copy number variants (CNVs) [
30].
Unique molecular identifiers (UMIs) are often used to barcode individual templates so that the amplicons from different templates can be identified [
31]. UMIs are normally short DNA fragments that are constituted by random nucleotide bases. The idea is that for a DNA sequence of N bases, there will be 4N different types of barcodes that can be used to label different templates before amplification. If a mutation does not appear in most of the same UMI-connected sequences, it is likely to be an error introduced by amplification or the sequencing reaction. By using this strategy, the Safe-SeqS reduces the sequencing errors by at least 70-fold and has a sensitivity as high as ~98% for detecting tumor-specific mutations [
32].
A combination of optimized library preparation and bioinformatics algorithm is often used to achieve better performance of NGS assays for low-abundance sequence aberration in ctDNA analysis. Target enrichment of DNA fragments containing recurrent mutations combined with deep sequencing can also improve the sensitivity of mutation detection. CAncer Personalized Profiling by deep Sequencing (CAPP-Seq) is such an NGS assay that combines a target-selection library preparation approach with specialized bioinformatics workflow [
33]. It generates a library of selectors that can specifically bind to recurrently mutated genomic regions for target enrichment. Combined with dedicated bioinformatics analysis, CAPP-Seq can detect MAF ~ 0.02% with a sensitivity of nearly 100% among stage II-IV NSCLC patients [
33]. Targeted error correction sequencing (TEC-Seq) is another example of experimental and bioinformatic optimizations to improve assay performance, including optimized target capture and library preparation, maximizing the representation of unique cfDNA both using different mapping positions and molecular barcodes, redundant sequencing, filtering of mapping and sequencing artefacts, and identifying and removing normal cell proliferation alterations [
34]. Target capturing a panel of genes and deep sequencing (~30,000×) of the captured DNA fragments is performed to ensure high sensitivity and specificity. It was able to detect somatic mutations in the plasma of 71%, 59%, 59% and 68%, respectively in colorectal, breast, lung, or ovarian cancer patients with stage I or II diseases, demonstrating very promising results for broad cancer diagnosis at early stages.
This entry is adapted from the peer-reviewed paper 10.3390/life11090890