In biosciences and biotechnologies, it is recently critical to promote research regarding the regulation of the dynamic functions of proteins of interest. Light-induced control of protein activity is a strong tool for a wide variety of applications because light can be spatiotemporally irradiated in high resolutions. Therefore, synthetic, semi-synthetic, and genetic engineering techniques for photoactivation of proteins have been actively developed. As a solution for overcoming barriers in conventional ones, researchers' recent approaches in which proteins were chemically modified with biotinylated caging reagents are introduced to photo-activate a variety of proteins without genetic engineering and elaborate optimization.
- protein caging
- optogenetics
- genetic code extension technology
- expressed protein ligation
- photolytic protein aggregates
1. Introduction
2. Caged Proteins
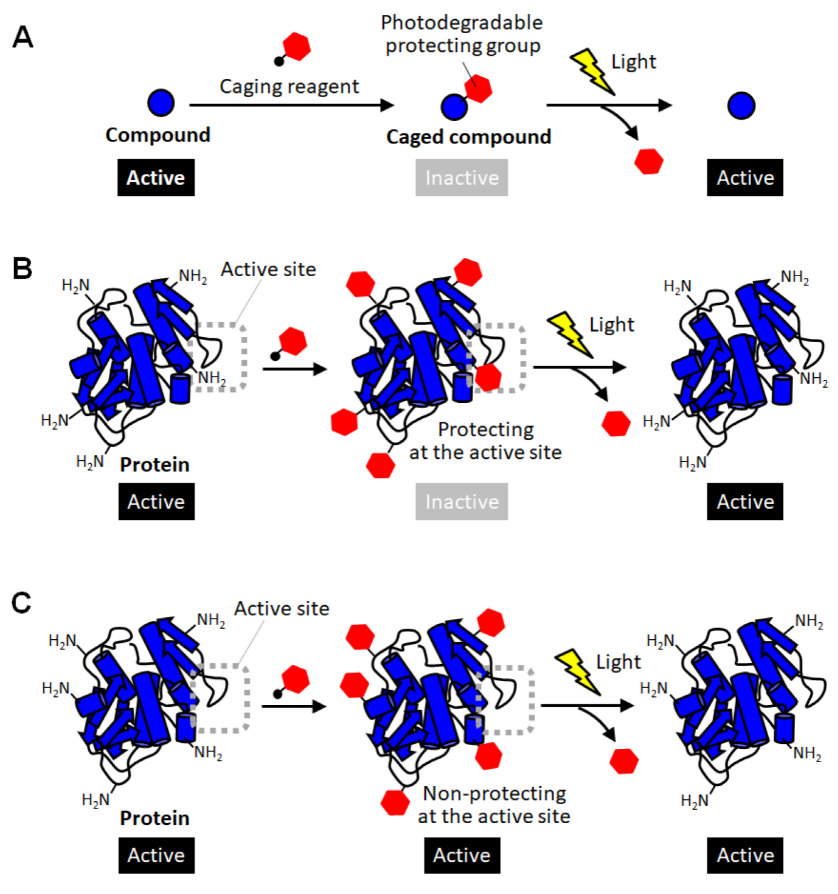
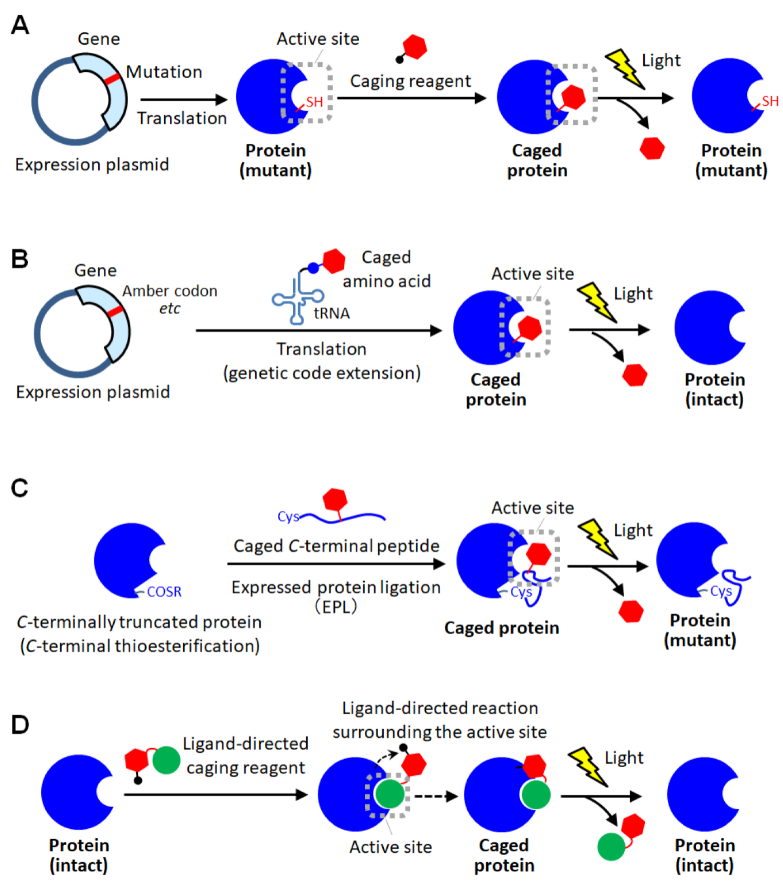
3. Site-Specific Protein Caging
This entry is adapted from the peer-reviewed paper 10.3390/app12083750
References
- Gautier, A.; Gauron, C.; Volovitch, M.; Bensimon, D.; Jullien, L.; Vriz, S. How to control proteins with light in living systems. Nat. Chem. Biol. 2014, 10, 533–541.
- Wu, Y.I.; Frey, D.; Lungu, O.I.; Jaehrig, A.; Schlichting, I.; Kuhlman, B.; Hahn, K.M. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 2009, 461, 104–108.
- Inagaki, H.K.; Jung, Y.; Hoopfer, E.D.; Wong, A.M.; Mishra, N.; Lin, J.Y.; Tsien, R.Y.; Anderson, D.J. Optogenetic control of Drosophila using a red-shifted channel rhodopsin reveals experience-dependent influences on courtship. Nat. Methods 2014, 11, 325–332.
- Morri, M.; Sanchez-Romero, I.; Tichy, A.M.; Kainrath, S.; Gerrard, E.J.; Hirschfeld, P.P.; Schwarz, J.; Janovjak, H. Optical functionalization of human Class A orphanG-protein-coupled receptors. Nat. Commun. 2018, 9, 1950.
- Wang, J.; Liu, Y.; Liu, Y.; Zheng, S.; Wang, X.; Zhao, J.; Yang, F.; Zhang, G.; Wang, C.; Chen, P.R. Time-resolved protein activation by proximal decaging in living systems. Nature 2019, 569, 509–513.
- Tomatsu, I.; Peng, K.; Kros, A. Photoresponsive hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2011, 63, 1257–1266.
- Høgset, A.; Prasmickaite, L.; Selbo, P.K.; Hellum, M.; Engesæter, B.Ø.; Bonsted, A.; Berg, K. Photochemical internalisation in drug and gene delivery. Adv. Drug Deliv. Rev. 2004, 56, 95–115.
- Gu, Z.; Biswas, A.; Zhao, M.; Tang, Y. Tailoring nanocarriers for intracellular protein delivery. Chem. Soc. Rev. 2011, 40, 3638–3655.
- Sasaki, Y.; Akiyoshi, K. Nanogel engineering for new nanobiomaterials: From chaperoning engineering to biomedical applications. Chem. Rec. 2010, 10, 366–376.
- Brieke, C.; Rohrbach, F.; Gottschalk, A.; Mayer, G.; Heckel, A. Light-controlled tools. Angew. Chem. Int. Ed. 2012, 51, 8446–8476.
- Ghosh, M.; Song, X.; Mouneimne, G.; Sidani, M.; Lawrence, D.S.; Condeelis, J.S. Cofilin promotes actin polymerization and defines the direction of cell motility. Science 2004, 304, 743–746.
- Deiters, A.; Groff, D.; Ryu, Y.; Xie, J.; Schultz, P.G. A genetically encoded photocaged tyrosine. Angew. Chem. Int. Ed. 2006, 45, 2728–2731.
- Selbo, P.K.; Sandvig, K.; Kirveliene, V.; Berg, K. Release of gelonin from endosomes and lysosomes to cytosol by photochemical internalization. Biochim. Biophys. Acta 2000, 1475, 307–313.
- Peng, K.; Tomatsu, I.; Kros, A. Light controlled protein release from a supramolecular hydrogel. Chem. Commun. 2010, 46, 4094–4096.
- Levskaya, A.; Weiner, O.D.; Lim, W.A.; Voigt, C.A. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 2009, 461, 997–1001.
- Kim, N.; Kim, J.M.; Lee, M.; Kim, C.Y.; Chang, K.Y.; Heo, W.D. Spatiotemporal control of fibroblast growth factor receptor signals by blue light. Chem. Biol. 2014, 21, 903–912.
- Nihongaki, Y.; Kawano, F.; Nakajima, T.; Sato, M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 2015, 33, 755–760.
- Gil, A.A.; Carrasco-López, C.; Zhu, L.; Zhao, E.M.; Ravindran, P.T.; Wilson, M.Z.; Goglia, A.G.; Avalos, J.L.; Toettcher, J.E. Optogenetic control of protein binding using light-switchable nanobodies. Nat. Commun. 2020, 11, 4044.
- Zhang, W.; Lohman, A.W.; Zhuravlova, Y.; Lu, X.; Wiens, M.D.; Hoi, H.; Yaganoglu, S.; Mohr, M.A.; Kitova, E.N.; Klassen, J.S.; et al. Optogenetic control with a photocleavable protein, Phocl. Nat. Methods 2017, 14, 391–394.
- Lu, X.; Wen, Y.; Zhang, S.; Zhang, W.; Chen, Y.; Shen, Y.; Lemieux, M.J.; Campbell, R.E. Photocleavable proteins that undergo fast and efficient dissociation. Chem. Sci. 2021, 12, 9658–9672.
- Ellis-Davies, G.C.R. Caged compounds: Photorelease technology for control of cellular chemistry and physiology. Nat. Methods 2007, 4, 619–6289.
- Curley, K.; Lawrence, D.S. Light-activated proteins. Curr. Opin. Chem. Biol. 1999, 3, 84–88.
- Lawrence, D.S. The preparation and in vivo applications of caged peptides and proteins. Curr. Opin. Chem. Biol. 2005, 9, 570–575.
- Matsuzaki, M.; Ellis-Davies, G.C.; Nemoto, T.; Miyashita, Y.; Iino, M.; Kasai, H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 2001, 4, 1086–1092.
- Marriott, G. Caged protein conjugates and light-directed generation of protein activity: Preparation, photoactivation, and spectroscopic characterization of caged G-actin conjugates. Biochemistry 1994, 33, 9092–9097.
- Thompson, S.; Spoors, J.A.; Fawcett, M.-C.; Self, C.H. Photocleavable nitrobenzyl–protein conjugates. Biochem. Biophys. Res. Commun. 1994, 201, 1213–1219.
- Self, C.H.; Thompson, S. Light activatable antibodies: Models for remotely activatable proteins. Nat. Med. 1996, 2, 817–820.
- Ottl, J.; Gabriel, D.; Marriott, G. Preparation and photoactivation of caged fluorophores and caged proteins using a new class of heterobifunctional, photocleavable cross-linking reagents. Bioconj. Chem. 1998, 9, 143–151.
- Bédouet, L.; Adenier, H.; Pulvin, S.; Bedel-Cloutour, C.; Thomas, D. Recovery of the oxidative activity of caged bovine haemoglobin after UV photolysis. Biochem. Biophys. Res. Commun. 2004, 320, 939–944.
- Marriot, G.; Heidecker, M. Light-directed generation of the actin activated ATPase activity of caged heavy meromysin. Biochemistry 1996, 35, 3170–3174.
- Golan, R.; Zehavi, U.; Naim, M.; Patchornik, A.; Smirnoff, P.; Inhibition of, E. coli β-galactosidase by 2-nitro-1-(4,5-dimethoxy-2-nitrophenyl)ethyl, a photo-reversible thiol label. Biochim Biophys Acta 1996, 1293, 238–242.
- Zou, K.; Cheley, S.; Givens, R.S.; Bayley, H. Catalytic subunit of protein kinase A caged at the activating phosphothreonine. J. Am. Chem. Soc. 2002, 124, 8220–8229.
- Ghosh, M.; Ichetovkin, I.; Song, X.; Condeelis, J.S.; Lawrence, D.S. A new strategy for caging proteins regulated by kinases. J. Am. Chem. Soc. 2002, 124, 2440–2441.
- Chang, C.Y.; Niblack, B.; Walker, B.; Bayley, H. A photogenerated pore-forming protein. Chem. Biol. 1995, 2, 391–400.
- Mendel, D.; Eilman, J.A.; Schultz, P.G. Construction of a light-activated protein by unnatural amino acid mutagenesis. J. Am. Chem. Soc. 1991, 113, 2758–2760.
- Cook, S.N.; Jack, W.E.; Xiong, X.; Danley, L.E.; Ellman, J.A.; Schultz, P.G.; Noren, C.J. Photochemically lnitiated Protein Splicing. Angew. Chem. Int. Ed. 1995, 34, 1629–1630.
- England, P.M.; Lester, H.A.; Davidson, N.; Dougherty, D.A. Site-specific, photochemical proteolysis applied to ion channels in vivo. Proc. Natl. Acad. Soc. USA 1997, 94, 11025–11030.
- Miller, J.C.; Silverman, S.K.; England, P.M.; Dougherty, D.A.; Lester, H.A. Flash decaging of tyrosine sidechains in an ion channel. Neuron 1998, 20, 619–624.
- Short, G.F.; Lodder, M.; Laikhter, A.L.; Arslan, T.; Hecht, S.M. Caged HIV-1 protease: Dimerization is independent of the ionization state of the active site aspartates. J. Am. Chem. Soc. 1999, 121, 478–479.
- Tong, Y.; Brandt, G.S.; Li, M.; Shapovalov, G.; Slimko, E.; Karschin, A.; Dougherty, D.A.; Lester, H.A. Tyrosine decaging leads to substantial membrane trafficking during modulation of an inward rectifier potassium channel. J. Gen. Physiol. 2001, 117, 103–118.
- Petersson, E.J.; Brandt, G.S.; Zacharias, N.M.; Dougherty, D.A.; Lester, H.A. Caging proteins through unnatural amino acid mutagenesis. Methods Enzymol. 2003, 360, 258–273.
- Wu, N.; Deiters, A.; Cropp, T.A.; King, D.; Schultz, P.G. A genetically encoded photocaged amino acid. J. Am. Chem. Soc. 2004, 126, 14306–14307.
- Endo, M.; Nakayama, K.; Kaida, Y.; Majima, T. Design and synthesis of photochemically controllable caspase-3. Angew. Chem. Int. Ed. 2004, 116, 5761–5763.
- Endo, M.; Nakayama, K.; Majima, T. Design and synthesis of photochemically controllable restriction endonuclease BamHI by manipulating the salt-bridge network in the dimer interface. J. Org. Chem. 2004, 69, 4292–4298.
- Cornish, V.W.; Mendel, D.; Schultz, P.G. Probing protein structure and function with an expanded genetic code. Angew Chem Int Ed 1995, 34, 621–633.
- Rothman, D.M.; Petersson, E.J.; Vázquez, M.E.; Brandt, G.S.; Dougherty, D.A.; Imperiali, B. Caged phosphoproteins. J. Am. Chem. Soc. 2005, 127, 846–847.
- Ren, W.; Ji, A.; Ai, H. Caging proteins through unnatural amino acid mutagenesis. J. Am. Chem. Soc. 2015, 137, 2155–2158.
- Edwards, W.F.; Young, D.D.; Deiters, A. Light-activated cre recombinase as a tool for the spatial and temporal control of gene function in mammalian cells. ACS. Chem. Biol. 2009, 4, 441–445.
- Gautier, A.; Nguyen, D.P.; Lusic, H.; An, W.; Deiters, A.; Chin, J.W. Genetically encoded photocontrol of protein localization in mammalian cells. J. Am. Chem. Soc. 2010, 132, 4086–4088.
- Arbely, E.; Torres-Kolbus, J.; Deiters, A.; Chin, J.W. Photocontrol of tyrosine phosphorylation in mammalian cells via genetic encoding of photocaged tyrosine. J. Am. Chem. Soc. 2012, 134, 11912–11915.
- Lemke, E.A.; Summerer, D.; Geierstanger, B.H.; Brittain, S.M.; Schultz, P.G. Control of protein phosphorylation with a genetically encoded photocaged amino acid. Nat. Chem. Biol. 2007, 3, 769–772.
- Walker, O.S.; Elsässer, S.J.; Mahesh, M.; Bachman, M.; Balasubramanian, S.; Chin, J.W. Photoactivation of mutant isocitrate dehydrogenase 2 reveals rapid cancer-associated metabolic and epigenetic changes. J. Am. Chem. Soc. 2016, 138, 718–721.
- Hemphill, J.; Borchardt, E.K.; Brown, K.; Asokan, A.; Deiters, A. Optical Control of CRISPR/Cas9 Gene Editing. J. Am. Chem. Soc. 2015, 137, 5642–5645.
- Hiraoka, T.; Hamachi, I. Caged RNase: Photoactivation of the enzyme from perfect off-state by site-specific incorporation of 2-nitrobenzyl moiety. Bioorg. Med. Chem. Lett. 2003, 13, 13–15.
- Pellois, J.P.; Hahn, M.E.; Muir, T.W. Simultaneous triggering of protein activity and fluorescence. J. Am. Chem. Soc. 2004, 126, 7170–7171.
- Hahn, M.E.; Muir, T.W. Photocontrol of Smad2, a multiphosphorylated cell-signaling protein, through caging of activating phosphoserines. Angew. Chem. Int. Ed. 2004, 43, 5800–5803.
- Cotton, G.J.; Muir, T.W. Peptide ligation and its application to protein engineering. Chem. Biol. 1999, 6, R247–R256.
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1999, 266, 776–778.
- Tang, S.; Wan, Z.; Gao, Y.; Zheng, J.S.; Wang, J.; Si, Y.Y.; Chen, X.; Qi, H.; Liu, L.; Liu, W. Total chemical synthesis of photoactivatable proteins for light-controlled manipulation of antigen–antibody interactions. Chem. Sci. 2016, 7, 1891–1895.
- Curley, K.; Lawrence, D.S. Photoactivation of a signal transduction pathway in living cells. J. Am. Chem. Soc. 1998, 120, 8573–8574.
- Loudwig, S.; Nicolet, Y.; Masson, P.; Fontecilla-Camps, J.C.; Bon, S.; Nachon, F.; Goeldner, M. Photoreversible inhibition of cholinesterases: Catalytic serine-labeled caged butyrylcholinesterase. ChemBioChem 2003, 4, 762–767.
- Tsukiji, S.; Miyagawa, M.; Takaoka, Y.; Tamura, T.; Hamachi, I. Ligand-directed tosyl chemistry for protein labeling in vivo. Nat. Commun. 2008, 5, 341–343.
- Fujishima, S.; Yasui, R.; Miki, T.; Ojida, A.; Hamachi, I. Ligand-directed acyl imidazole chemistry for labeling of membrane-bound proteins on live cells. J. Am. Chem. Soc. 2012, 134, 3961–3964.
- Tamura, T.; Ueda, T.; Goto, T.; Tsukidate, T.; Shapira, Y.; Nishikawa, Y.; Fujisawa, A.; Hamachi, I. Rapid labelling and covalent inhibition of intracellular native proteins using ligand-directed N-acyl-N-alkyl sulfonamide. Nat. Commun. 2018, 14, 1870.
- Matsuo, K.; Kioi, Y.; Yasui, R.; Takaoka, Y.; Miki, T.; Fujishima, S.; Hamachi, I. One-step construction of caged carbonic anhydrase I using a ligand-directed acyl imidazole-based protein labeling method. Chem. Sci. 2013, 4, 2573–2580.