2. Caged Proteins
Methods for converting proteins to photo-responsive ones by reversible chemical modification have also been used as a complementary approach in which intact proteins with no modification are produced after photoactivation [
10,
11,
12,
21,
22,
23]. In the field of chemical biology, inactivation of biomolecules by modification with photodegradable protective groups is called “caging”, and such protected compounds are called caged compounds [
10,
21]. Caging have been employed for activating biomolecules of interest at desired timing and location by exposure to light (
Figure 1A). For example, a caged glutamine was reported to be introduced into a neuron, and then one of the dendrites was site-specifically exposed to light. As a result, the shape change of the light-exposed dendrites clearly indicated the molecular system in which glutamine affects the shape memory of neural spines [
24]. Thus, caging has been utilized as a powerful research tool for giving new insight into molecular mechanisms in cell biology. Such a caging approach has also been applied to proteins, and caged proteins have been reported for more than a quarter of a century [
22,
23]. The simplest strategy for preparing caged proteins is to randomly introduce a photodegradable protecting group to the amino group on the protein surface through the reaction with an active carbonyl group (
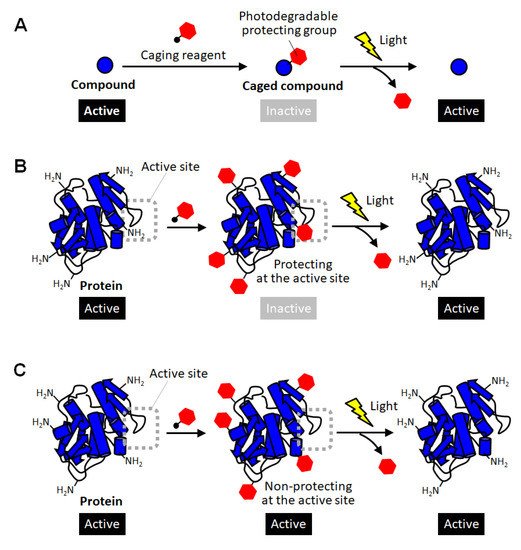
Figure 1B) [
25,
26,
27,
28]. In this method, in principle, almost all proteins can be caged without any pretreatment simply by mixing a reactive photodegradable protecting reagent (a caging reagent). After the protective group is degraded by light, the caged lysine residue returns to the original lysine one without leaving any trace, so there is no need to consider the effect of caging on the protein function after photoactivation. This is a great advantage when studying the function of a protein or using it as a drug. Actually, the polymerization activity of G-actin was photo-regulated by this caging strategy [
25]. The selective binding function of antibodies were demonstrated to be remotely controlled by light exposure [
27]. In a similar way, the carboxylic acid moiety on protein surfaces was reported to be randomly blocked with the diazo derivatives of photolytic protection groups [
29]. This method realized photoactivation of hemoglobin. However, unlike small caged compounds, such random introduction of a small photodegradable protecting group onto the surface of a large protein often fails to fully inactivate the function of the protein (
Figure 1C). In the case of enzymes, for example, this simple strategy based on random amine modification can function only on the enzymes that happen to have lysine residues at sites involved in catalytic activity or substrate binding (
Figure 1B). Therefore, caging of proteins often requires the strategy of introducing the photodegradable protecting group specifically at the sites involved in their activity.
Figure 1. Schematic illustration of caging of compounds and proteins. (A) Caging of small molecular compounds. (B) Successful and (C) unsuccessful caging of proteins through random modification of lysine residues.
3. Site-Specific Protein Caging
As a method for site-specific introduction of the photodegradable protecting group, the use of the cysteine residue of the proteins has been utilized [
11,
30,
31,
32,
33,
34]. The selective reactivity of the thiol group realized the site-specific caging of the cysteine residue of which there are often one or a few on protein surfaces. Similar to the random amine coupling, the function of intact proteins was reported to be photo-regulated by caging with thiol-reactive protecting reagents when the cysteine residues were at the sites involved in protein functions [
30,
31]. By this approach, the enzymatic activities of myosin [
30] and β-galactosidase [
31] were demonstrated to be activated by light exposure. As another method using the thiol-based reaction, the caging of thiophosphorylated proteins was also reported [
32]. In this method, the threonine residue of a protein was thiophosphorylated using 3-phosphoinositide-dependent kinase, and then modified with a thiol-reactive caging reagent. After photolysis, the caged protein converted to the thiophosphorylated protein which exerted almost the same activity as the phosphorylated one. This kinase-coupled approach may be versatile for caging a variety of phosphorylated proteins. To apply such thiol-based site-specific caging approaches to any other proteins, the use of cysteine-substituted mutants has been widely utilized (
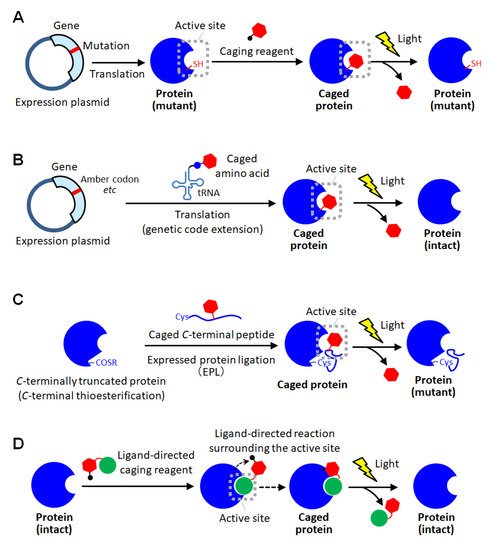
Figure 2A) [
11,
33,
34]. In this approach, the function-related site is replaced with cysteine, and a caging reagent that selectively reacts with the thiol group is applied. Based on this approach, the actin polymerization activity of cofilin, which was unknown so far, was exerted by light exposure in living cells, giving new insights into the role of cofilin [
11]. Furthermore, light-induced depolymerization of actin was also achieved by caged cofilin on the glass substrate [
33], and the pore-formation on cellular membranes was photo-induced by caged α-hemolysin [
34]. Although this approach is simple to introduce a protecting group at the desired active site, the proteins produced after photolysis are only the cysteine-substituted mutant protein on which the mutation is introduced at a critical site involved in function (
Figure 2A). Therefore, there is a high risk that the light-induced activity of the mutant protein will be inferior to that of the native one. Thus, the caging strategy based on post-translational chemical modification often requires the site-specific replacement with reactive amino acids for selective caging, and therefore, almost exclusively applied to mutant proteins.
Figure 2. Schematic illustration of site-specific caging of proteins. (A) Caging based on cysteine mutagenesis. (B) Caging based on unnatural amino acid introduction using genetic code extension technologies. (C) Caging based on protein semi-synthesis using expressed protein ligation (EPL). (D) Caging based on ligand-directed chemistry.
As a method for site-specific protein caging, another genetic approach using unnatural caged amino acids has been widely employed [
5,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52,
53]. Using the genetic code extension technology such as notably amber mutation and four-base codon mutation [
43], in which an unnatural amino acid is coded to a specific codon, the desired position of the protein of interest can be replaced with a caged amino acid (
Figure 2B). Until now, caged aspartic acid [
35,
39], caged serine [
36,
51], caged glycine [
37,
43], caged tyrosine [
5,
38,
40,
48,
50], caged cysteine [
42,
47], caged lysine [
44,
49,
52,
53], caged phosphoserine [
46] and caged phosphotyrosine [
46] have been utilized for photo-activation of various proteins including enzymes [
35,
39,
42,
43,
44,
45], intein proteins [
36,
47], ion channels [
37,
38,
40], receptors [
46], phosphorylation cascade proteins [
50,
51], and so on [
5,
48,
49,
52,
53]. The site-specific incorporation of unnatural caged amino acid into proteins was first achieved in
E.
coli [
35,
36,
42,
43,
44] and a cell-free protein expression system [
46], and subsequently their photoactivation was examined in vitro. Furthermore, the recombinant caged proteins were transduced into mammalian cells with a transfection reagent to utilize their photoactivatable functions for clarifying the spatial and temporal molecular mechanism of the living system [
5,
48]. Next, to photo-regulate ion channels on the plasma membrane of living cells, in situ expression of site-specifically caged proteins was performed in
Xenopus oocytes by injecting the caged amino acids-combined tRNA through microinjection [
37,
38,
40]. Recently, the genetic code extension technology has become applicable to eukaryotic cells by using the genetic expression system for the orthogonal pair of pyrrolysyl-tRNA synthetase and the corresponding tRNA, enabling the genetic incorporation of caged amino acids into proteins in yeast cells [
42,
51] and mammalian cells [
5,
49,
50,
51,
52,
53]. By using such a genetic encoding system, a pioneer group, Chin et al. demonstrated light-induced intracellular localization change of caged nuclear localization proteins in human cells [
49] and achieved photoactivation of receptor-mediated signal transduction by caging a phosphorylation protein, STAT1 [
50]. Furthermore, to understand the role of cancer-specific mutation, they reported to photo-activate the synthesis of the oncometabolite (
R)-2-hydroxyglutarate through caging an isocitrate dehydrogenase mutant in normal cells and exposing those engineered cells to light [
52]. Such genetic caging with unnatural amino acid introduction was also reported to achieve light-induced gene editing through caging of Cas9 [
53], photo-activation of immune response through caging of MEK1 [
5], and screening of caspase substrate proteins through caging of caspase-3 [
5] in human cells. Thus, in this approach, a variety of wild-type proteins could be site-specifically caged by utilizing various caged amino acids. Moreover, this approach is advantageous in introducing a photodegradable protection into the active site inside the closed structure of proteins because the caged amino acid can be incorporated on translation before protein folding. However, it requires advanced and specialized gene engineering techniques, and therefore is not easily available.
Other strategies in protein caging have also been reported. In semisynthetic approaches, a small part of the protein was chemically synthesized by incorporating caged amino acid through peptide synthesis, and then linked with a major part of the protein to produce a site-specifically caged protein [
54,
55,
56]. First, as a pioneering approach, site-specific caged ribonuclease S (RNase S) was prepared by mixing a synthesized caged S-peptide and S-protein [
54]. RNase S is consisting of two peptide fragments, S-peptide (1-20 residues) and S-protein (21-124 residues), and these fragments bind in a self-assembly manner to exert the enzymatic activity. In this report, by optimizing the replacement site of caged amino acids in S-peptide, the caged RNase S was activated by light in an off-on manner [
54]. This approach was greatly developed by the expressed protein ligation (EPL) method (
Figure 2C) [
55,
56]. In EPL-based caging, the fusion protein of a
C-terminally-truncated target protein with a self-processing intein domain is overexpressed in
E.
coli, and through the intein-mediated processing reaction [
57], the
C-terminal is converted to a reactive thioester moiety. In parallel, the
C-terminal domain peptide including a cysteine residue at the
N-terminal end and caged amino acids at the desired positions is chemically synthesized. Then, the caged
C-terminal peptide is linked to the
C-terminal end of the truncated protein through native chemical ligation [
58]. A signal transduction protein with multiple caged phosphorylated serine residues was reported to be prepared by this approach [
55,
56]. Based on the high degree of certainty and freedom of peptide synthesis, the semisynthetic approaches allow for the precise introduction of multiple photodegradable protecting groups and the expansion of the range of amino acids that can be protected. However, the caging position is limited to the terminal sequence of proteins, leading to limitation of applicable proteins. Recently, a total chemical synthesis-based approach was also reported to overcome this limitation of the semisynthetic protein caging [
59], but it requires chemical reactions that are too specialized for anyone to immediately utilize. In ligand-directed caging approaches, a ligand molecule that selectively binds to the active site of a protein assists site-specific modification with the caging group [
60,
61]. In a pioneering study of this approach, the substrate peptide with a thiol-reactive moiety through a photocleavable linker was utilized for active-site specific caging [
60]. This synthesized peptide was recognized with enzyme and reacted with the cysteine residue selectively at the active site. Based on this approach, a protein kinase was caged in vitro and introduced into living cells by microinjection, leading to light-induced activation of phosphorylation for cellular morphological change [
60]. Similarly, a small substrate analogue with photodegradable ability was reported to specifically incorporated into a serine residue at the active site of enzyme, and the protected activity of enzyme was regenerated by light [
61]. Hamachi et al. have presented a number of sophisticated reports in ligand-directed chemistry for in situ protein labeling [
62,
63,
64]. For site-specific caging, a photocleavable linker was inserted between the ligand for proteins of interest and a special reactive group, and after mixing with proteins, this caging reagent could be selectively attached to amino acid residue surrounding the active site through a ligand-directed proximity effect on the reaction (
Figure 2D) [
65]. By employing this method, an enzyme and a receptor protein were efficiently caged with the reagents including specific ligand, respectively, and demonstrated to be uncaged by light exposure. Thus, ligand-directed approaches can achieve site-specific caging of intact proteins of interest simply by mixing with the specific caging reagents. However, the application is limited to enzymes and binding proteins. Moreover, ligands with an appropriate dissociation constant, which must meet both the requirements for the selective binding in the caging step and the rapid releasing in the photoactivation step, are needed. Such demands of this approach may be issues in terms of versatility.