Cdc25 phosphatases have been considered promising targets for anticancer development due to the correlation of their overexpression with a wide variety of cancers. In the past decades, the interest in this subject has considerably increased and many publications have been launched concerning this issue.
1. Introduction
The cell division cycle 25 (Cdc25) phosphatases are dual-specificity phosphatases (DSPs) that catalyze the dephosphorylation of the Cdk/Cyc protein complex, an important regulator of the human cell cycle [
1]. In human cells, three Cdc25 phosphatases are characterized, Cdc25A, Cdc25B, and Cdc25C, which share the similarity of amino acids identity from 20 to 25% for N-terminal and 60% similarity for C-terminal, and are differentially expressed in the cell division cycle [
2,
3].
Cdc25A is involved in the control of the G1/S transition by dephosphorylating and thus activating Cdk2/cyclin E and cyclin A complexes, as well as controlling the progression into mitosis. Additionally, Cdc25A activates Cdk1/cyclin B provoking the transition G
2/M [
4], while Cdc25B and C mainly regulate the progression at the G2/M transition and mitosis [
4,
5]. Cdc25B accumulates from the late S and early G2 phases of the cell cycle and its activity peaks at the G2/M transition [
4,
5]. Cdc25B is proposed to play a “starter” role in triggering mitosis by dephosphorylation and thereby activating Cdk1/cyclin B at the centrosome level [
4,
5]. On the other hand, Cdc25C is mainly involved in controlling progression into mitosis and this activation is correlated to Cdc25C phosphorylation by Cdk1/cyclinB substrate [
5]. The Cdc25 phosphatases also play a crucial role in the checkpoint response that prevents Cdk/cyclin activation following DNA damage [
6,
7].
Many studies have shown that Cdc25 is highly expressed in cancer. Therefore, it is considered an excellent target for cancer therapy [
5,
8,
9]. Thus, inhibition of these phosphatases may represent a promising therapeutic approach in oncology [
10,
11,
12].
This entry is dedicated to searching for safe and efficient Cdc25s inhibitors derived from natural, synthetic, and computational sources.
2. Structure of Cdc25 Phosphatase
The Cdc25 proteins are 450–600 residues in length and divided into two regions, the N-terminal, and C-terminal regions. The N-termini are highly divergent in sequence because of the alternative splicing therein. They contain numerous sites for phosphorylation and ubiquitination involved in regulating phosphatase activity [
6]. The C-terminal regions (~60% pairwise identity) contain the catalytic functionality of the Cdc25s. The signature motif (HCX
5R), containing the catalytic cysteine that defines the active sites of all protein tyrosine phosphatases (PTPs), represents the only significant region of homology between the Cdc25s and other PTPs. Motif C is the catalytic cysteine, while the five X residues form a loop whose backbone amides a hydrogen bond to the phosphate of the substrate. Finally, the motif R is a highly conserved arginine that is required for binding and transition-state stabilization of the phosphate [
6].
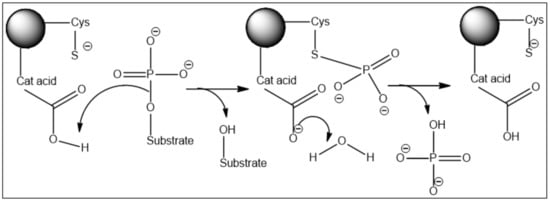
3. Mechanism of Dephosphorylation of Cdc25 Substrate
Like other dual-specificity phosphatases (DSPs), Cdc25s have the ability to hydrolyze phosphoserine/threonine as well as phosphotyrosine residues. Cdc25B-catalyzed dephosphorylation of protein substrates proceeds via a mechanism using a monoprotonated phosphate with an apparent pKa of 6.5. This mechanism is a pH-dependent reaction. Accordingly, at high pH values, the rate of the reaction is slower than at low pH values [
13].
The active site contains two key catalytic residues, an unprotonated thiolate and a protonated catalytic acid. The phosphoryl group of the substrate is cradled by the active site loop, forming hydrogen bonds to the backbone amides and the arginine of the HCX
5R motif. The thiolate anion of the catalytic cysteine lies directly below the phosphoryl group, which sets up an in-line attack of the thiolate on the P-O bond in the first chemical step of the reaction. The departure of the leaving group and formation of the phospho-cysteine intermediate are facilitated by the protonation of the oxyanion by the catalytic acid (still unknown) located in a separate loop. In the second chemical step of the reaction, water serves as a nucleophile in the hydrolysis of the phospho-enzyme intermediate assisted by the aspartate (
Figure 1) [
6].
Figure 1. Two-step reaction mechanism of the Cdc25 phosphatases with a covalent phospho-cysteine intermediate.
It is noteworthy that the thiolate of the active cysteine (Cys473 in Cdc25B) is found to have an extremely high rate of conversion to sulfenic acid. When it is tested with hydrogen peroxide, the result for Cdc25B is 15-fold and 400-fold faster than that for the protein tyrosine phosphatase PTP1B and the cellular reductant glutathione, respectively. This suggests that Cys426 has a preventing role in the formation of sulfenic acid (Cys-SO
2) in the Cdc25s by forming a rapid intramolecular disulfide linkage with catalytic cysteines (Cys473). This intramolecular disulfide is reversibly and rapidly reduced by cellular thioredoxin/thioredoxin reductase. Thus, the chemistry and kinetics of the active-site cysteines of the Cdc25s support a physiological role for reversible redox-mediated regulation of the Cdc25s as important regulators of the eukaryotic cell cycle [
14].
4. Cdc25 Phosphatases and Cancer
The Cdc25 protein family plays a crucial role in controlling cell proliferation, making it an excellent target for cancer therapy [
5,
8,
9,
15]. Cdc25A and Cdc25B are over-expressed in many different primary human cancers [
6,
16] in the following percentages, respectively: breast cancer: 70% and 57%, thyroid cancer: 17–69% and 36–64%, hepatocellular cancer: 56% and 20%, ovarian cancer: 30% for both of them, colorectal cancer: 47–53% and 43–67%, non-Hodgkin’s lymphoma: 50% and 65%, laryngeal cancer: 41% and 57% and oesophageal cancer: 46–66% and 48–79% [
17]. Cdc25B is overexpressed solely in Gastric (78%) endometrial (73%) and prostate (30%) cancers and Gliomas (47%). The incidence of overexpression of Cdc25C has been barely reported [
17]. In addition, Cdc25A and Cdc25B are overexpressed together in 30% of tumors while, in 45% of them, one only is overexpressed [
17].
5. Molecular Studies of Cdc25 Phosphatase
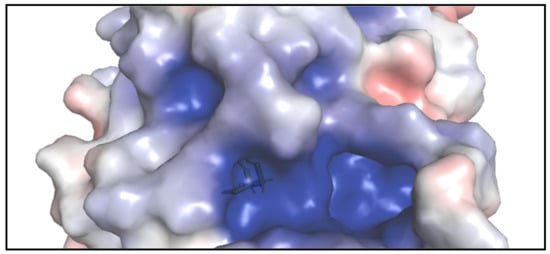
The crystal structure of the catalytic domain of Cdc25B displays a well-ordered active site containing a bounded sulfate. This active site loop creates a special environment for cysteine 473 [
18,
19]. Despite the lack of crystallographic structure in the protein–inhibitor complex and the nonexistence of a deep active site pocket, one of the largest cavities on the surface of Cdc25B adjacent 0.6 Å to the active site reveals good binding envelop for the small inhibitory molecules during docking processes (
Figure 2) [
18]. The residues that surround the inhibitor binding pocket adjacent to the active site contain Arg482, Arg544, and Thr547 besides other residues [
19].
Figure 2. The surface of Cdc25B shows the shallow active site (where the stick of Cys473 is shown) and the adjacent inhibitory pocket which is deeper and larger.
6. Interaction between Cdc25 Phosphatase and Its Protein Substrate
Several investigations have tried to predict a docked orientation for Cdc25B with its Cdk2-pTpY-CycA protein substrate using different docking techniques [
20]. The available crystal structure containing a bound sulfate (1QB0) at the active site is expected to be a good representative of the phosphate group of the phosphorylated protein substrate [
21]. However, on the Cdk2-pTpY-CycA substrate, the exact orientation of the
β-hairpin loop encompassing the adjacent Thr14 and Tyr15 sites of phosphorylation is unknown. The second point of contact between Cdc25B and its protein substrate is hot-spot residues (Arg488 and Tyr497). They are >20 Å from the active site and expected to make a contact with either Cdk2 or cyclin A at the binding interface [
20].
Computational docking of Cdk2-pTpY-CycA to the enzyme followed by refinement using molecular dynamics was performed. In addition, validation of the docking using X-ray crystallography was applied to the crystal structure of the substrate-trapping mutant of Cdc25B [
22,
23]. The phosphate of pThr14 is cradled within the active site loop with numerous hydrogen bonds to the amides backbone including the side chain of Arg479 of the CX5R signature motif. This phosphate is additionally coordinated in the active site pocket by contributions from the amide backbone of Thr14 and the side chain of Arg36 from the substrate [
20].
Further studies have been conducted to clarify the exact model for enzyme–substrate interaction. Some of these studies used small molecules such as
p-nitrophenyl phosphate (
pNPP) as substrate and applied the recently developed QM/MM Minimum Free Energy Path method. This was intended to gain an understanding of the dephosphorylation mechanism [
1].
This entry is adapted from the peer-reviewed paper 10.3390/molecules27082389