1. Diarylethene Cyclization
In this approach, an appropriate diarylethene derivative is prepared, which is then cyclized, usually by photochemical methods, to obtain helicene; the diarylethene is generally obtained either by the Wittig reaction between a phosphonium salt and an aldehyde, or by Heck coupling through a vinyl derivative and an aryl halide. It is possible to devise the synthesis by incorporating starting materials containing the desired side groups.
The Wittig reaction is compatible with several functional groups, including multiple bonds, halogens, nitro, ether, ester, and acetal groups, which therefore may be present on the aldehyde moiety. The phosphorus ylide may contain multiple bonds and several functional groups; if it contains electron-withdrawing groups, it becomes more stable. This approach was adopted in many cases for carbohelicenes, for instance in [
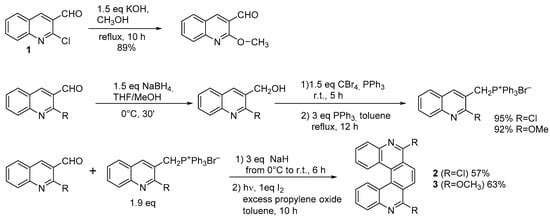
10], where 4-bromobenzaldehyde reacted with the appropriate four-ringed phosphonium ylide to obtain the diarylethene precursor of 2-Br-hexahelicene. For azahelicenes, the only additional limitation concerns the availability of the required commercial starting materials. One reported instance starts from 2-chloroquinoline-3-aldehyde (
1) to obtain the symmetrical 6,9-dichloro-(
2) and 6,9-dimethoxy-5,10-diaza[5]helicene (
3) [
11]. Both terms employed in the Wittig condensation originate from the same substituted quinolinaldehyde (
Scheme 1); (
1) is easily accessible by reacting acetanilide with POCl
3 in DMF at 80 °C [
12]. By using differently substituted moieties, it is also possible to obtain the unsymmetrically substituted product.
Scheme 1. Synthesis of 6,9-dichloro- and 6,9-dimethoxy-5,10-diaza[5]helicene.
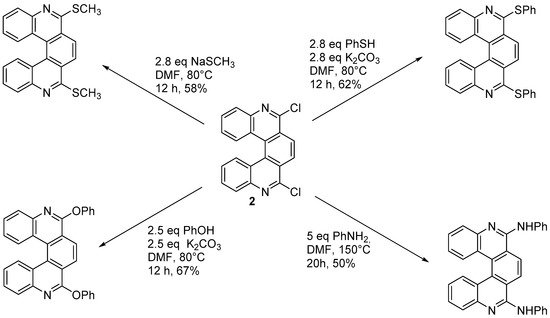
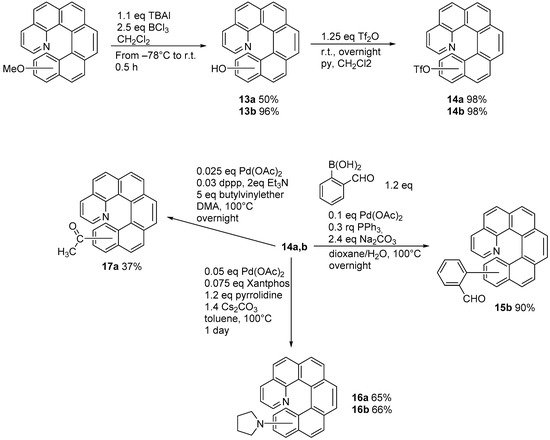
Starting from 6,9-dichloro-5,10-diaza[5]helicene (
2) it is possible to substitute chlorine atoms with several functional groups, as depicted in
Scheme 2.
Scheme 2. Introduction of functional groups starting from dichlorodiazahelicene 2.
The substitution of chlorine atoms with chiral amines provides a way to separate diastereomers, in order to achieve an enantiomeric resolution. However, the five-ringed helicenic scaffold is readily racemized, and the presence of nitrogen substitution accelerates racemization compared to the parent compound [
11].
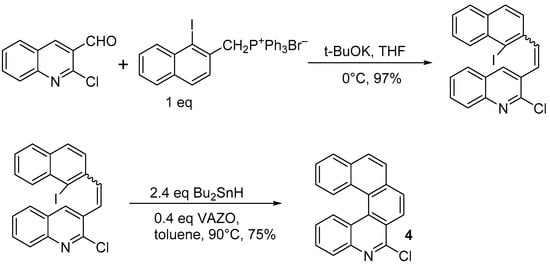
In a modification of this approach, Harrowven and coworkers [
13] prepared 6-chloro-5-aza[5]helicene (
4) again by using (
1) as the starting material, and reacting it with a phosphonium salt bearing an iodine atom, to be used for a non-photochemical radical coupling reaction (
Scheme 3).
Scheme 3. Synthesis of 6-chloro-5-aza[5]helicene 4 by radical cyclization.
3-bromo-4-aza[6]helicene was also prepared following the Wittig condensation. The photochemical ring closure strategy [
14] started from 6-bromo-2-pyridinecarboxaldehyde, which was condensed with benzo[g]phenanthren-3-ylmethyl triphenyl phosphonium bromide through the use of BuLi in THF to obtain the corresponding diarylethene in a 90% yield. This was then irradiated with visible light in toluene with I
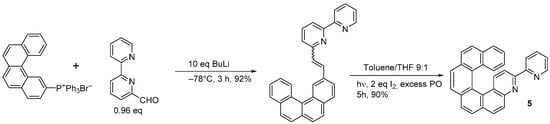
2 (2 equiv.) and excess propylene oxide (PO) for 15 h to yield 3-bromo-4-aza[6]helicene in a 45% yield. In the same way, racemic 3-(2-pyridyl)-4-aza[6]helicene
5 was synthesized in two steps from 2,2′-bipyridine-6-carbaldehyde by the Wittig reaction with (benzo[
g]phenanthren-3-ylmethyl)triphenyl phosphonium bromide followed by a photocyclization reaction (83% overall yield,
Scheme 4) [
15], and 3-(2-pyridyl)-4-aza[4]helicene was prepared by using (2-methylnaphthyl) triphenyl phosphonium bromide, with an overall yield of 61%.
Scheme 4. Synthesis of racemic 3-(2-pyridyl)-4-aza[6]helicene (5).
These compounds were used to realize Pt complexes acting as chiroptical switches. The 2,2′-bipyridine unit is endowed with remarkable stability and exceptional coordination chemistry. Functionalized helical bipyridine systems exhibit, in addition to the usual azahelicene properties, a rich and high-affinity coordination chemistry with metals, a good acid/base and alkylation reactivity, and redox activity.
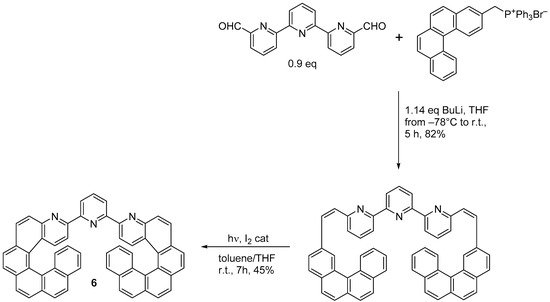
A chiroptical switch based on reversible Zn(II) complexation by the double azahelicene (
6) was synthesized by the same research group through double photochemical cyclization (
Scheme 5) [
16].
Scheme 5. Synthesis of 2,6-di[3-(4-aza[6]helicenyl)]pyridine (6).
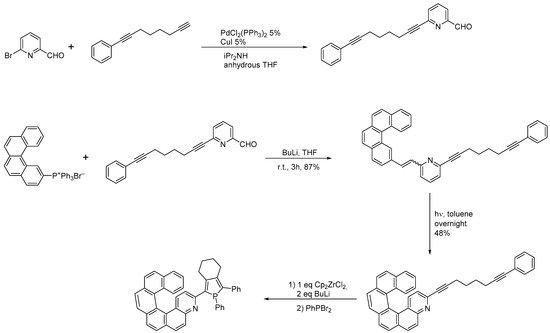
The same group also synthesized a three-substituted 4-aza[6]helicene by preparing a diarylethene already bearing a dialkynyl substituent, which, after cyclization, was then reacted further to obtain a phosphole-substituted azahelicene, as reported in
Scheme 6 [
17].
Scheme 6. Synthesis of a 4-aza[6]helicene bearing a phosphole moiety in position 3.
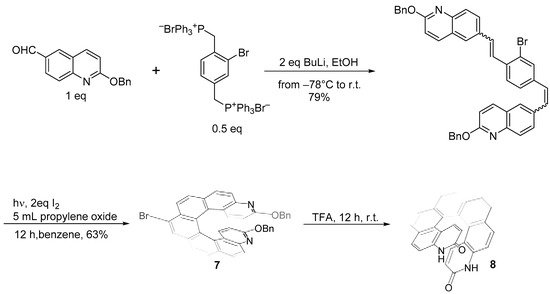
A Br-substituted diaza[7]helicene (
7) was prepared [
18] through the Wittig condensation, as shown in
Scheme 7.
Scheme 7. Synthesis of substituted 4,15-diaza[7]helicene 7.
Removal of the benzyloxy groups from (7) by TFA yields the corresponding brominated pyridinone 8 in a 63% yield. The bromine atom can be substituted with the COCH3 group. Pd- and CuI-cocatalyzed coupling of TMS-acetylene with 7 yielded the corresponding ethynyl helicene, which, by treatment with trifluoroacetic acid, cleanly liberated the pyridinone hydrogen bond sites and simultaneously deprotected the TMS-acetylene moiety, generating the methyl ketone derivative analogous to 8 in a nearly quantitative yield.
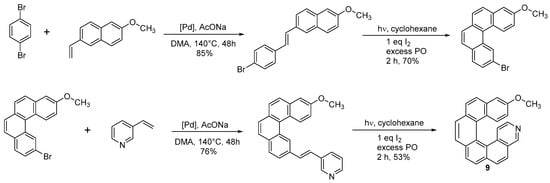
The same approach held for the Heck coupling, where both the alkene and the halogen-bearing moieties used to obtain the diarylethene could bear substituents. 14-methoxy-3-aza[6]helicene (
9) was synthesized through a double Heck condensation-photochemical oxidation approach (
Scheme 8) [
19].
Scheme 8. Synthesis of 14-methoxy-3-aza[6]helicene 9.
The Heck couplings were obtained by using excess of the commercially available vinylderivatives in the presence of 3 equiv. AcONa and 1% Hermann’s palladacycle [trans-di(μ-acetato)-bis[o-(di-o-tolylphosphino)-benzyl]dipalladium] as the catalyst in DMA. The -OCH3 group can be easily transformed in -OH by treatment with a BBr3 solution in CH2Cl2 at RT for 3 h, with an excellent yield. The hydroxy derivative could serve as N–O bidentate ligands in asymmetric synthesis or as building blocks for supramolecular architecture. Although not many instances have been described in the literature, this approach is quite general.
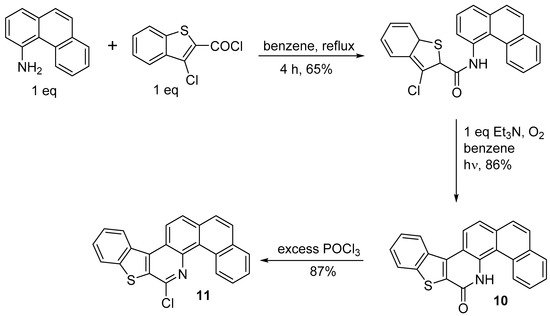
Only two successful syntheses of stilbenoid precursors by the photocyclization of amides have been described [
20]. In the first, photochemical ring closure of the amide provides lactam
10, which is then converted to double helicene
11 by POCl
3 (
Scheme 9).
Scheme 9. Synthesis of 8-chlorobenzo[4,5]thieno[2,3-c]naphtho[2,1-h]quinoline 11.
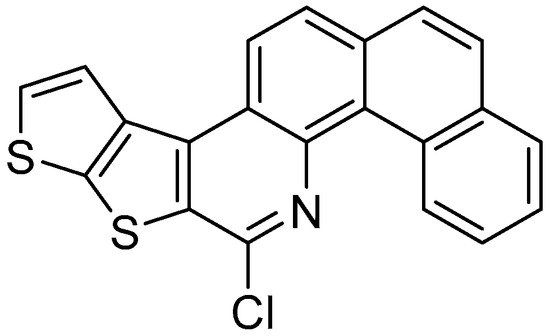
In the second, the same research group obtained the dithienocondensated aza[4]helicene (Figure 1) by the same procedure; however, the yield of the last step with POCl3 was moderate (39%).
Figure 1. 8-chloronaphtho[2,1-h]thieno[3’,2’:4,5]thieno[2,3-c]quinoline.
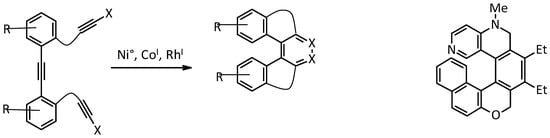
2. [2 + 2 + 2] Cycloisomerization
This approach to the synthesis of helicenes involves intramolecular [2 + 2 + 2] alkyne cycloisomerization catalyzed by Co
I, Ni
0, or Rh
I to provide the simultaneous formation of three or more cycles in a single step, giving access directly to the desired helicene, or to its tetrahydro derivative, to be oxidised in a subsequent step. This strategy has been extensively used for the synthesis of electron-rich systems, namely carbo- and oxa-helicenes [
21], while only limited examples are reported for electron-poor pyridine moieties (
Figure 2).
Figure 2. Scheme of the general [2 + 2 + 2] cycloisomerization reaction (left); a nitrogen-containing helicenoid structure obtained in this way (right).
Most frequently, unsubstituted azahelicenes are prepared by this method [
22]. Some attempts to apply this strategy to alkyl-substituted azahelicenes were only partially successful. Cycloisomerization of an azabiphenylylnaphthalene catalyzed by InCl
3 and PtCl
4 allowed for the synthesis of 6,10-dimethyl-2-aza-[6]helicene in an 80% yield [
23], while the same reaction, applied to the synthesis of 6,10-dimethyl-2,14-diaza-[6]helicene, failed despite the screening of a number of different catalyzers. In another instance [
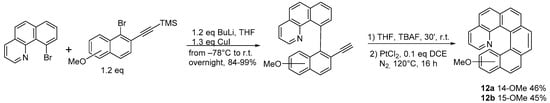
24], diversely substituted 1-aza[6]helicenes are produced in gram scale, starting from methoxy-substituted azahelicene (
12), obtained as in
Scheme 10.
Scheme 10. Synthesis of methoxy-substituted 1-aza[6]helicenes.
The methoxy group, which can be located either in position 14 (
12a) or 15 (
12b), can then be converted relatively easily and with fair to good yields into different functional groups (
Scheme 11). Furthermore, the precursors to compounds
12 show atropoisomeric chirality and could be resolved into stable enantiomers, thus enabling the obtainment of compounds
12a,
b with an enantiomeric excess of more than 90%.
Scheme 11. Transformations of functional groups onto the 1-aza[6]helicene framework.
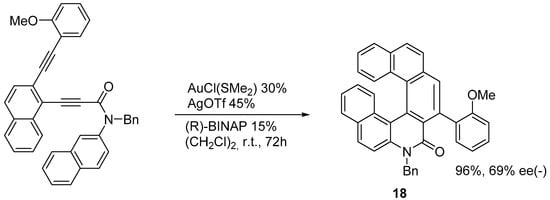
The enantioselective synthesis of azahelicenes and S-shaped double azahelicenes has been achieved via the Au-catalyzed sequential intramolecular hydroarylation of alkynes [
25]. By using the catalytic system indicated in
Scheme 12, the azahelicenone
18 is obtained in very good yield with 69% ee starting from an achiral precursor. It is also possible to reduce the amount of the catalyzers by operating at 80 °C: in this case, although, the yield decreases to 80%, and the ee decreases to 56%. When a phenyl group is introduced as a substituent in place of 2-methoxyphenyl, both the yield and ee decrease substantially.
Scheme 12. Preparation of a substituted 7-aza[6]helicenone.
Following the same procedure, products analogous to 18 can be prepared, in which one or both terminal aromatic rings bear two methoxy groups in positions 1,3.
Starting from a product analogous to 18 in which the nitrogen atom bears a 4-methoxybenzyl group, treatment with CF3COOH followed by reaction with POCl3 and PhNMe2 (-)-8-chloro-2-(2-methoxyphenyl)-7-aza[6]helicene 19 (Figure 3) obtained a 67% yield, with 74% ee. By using the same reaction strategy, it is also possible to obtain the S-shaped double azahelicene 20 represented in Figure 3, with >99% ee of the (+) enantiomer.
Figure 3. Azahelicenes obtained by sequential intramolecular Au-catalyzed diyne or tetrayne hydroarylation.
Double azahelicenes showed red shifts of absorption and emission maxima and higher quantum yields in the CHCl3 solution compared with azahelicenes. The optical rotation values of double azahelicenes were smaller than those of the corresponding single azahelicenes.
3. Other Synthetic Methods
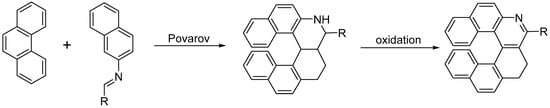
The Povarov reaction is a formal inverse-electron demand [4 + 2] cycloaddition between electron rich-alkenes, acting as dienophiles, and
N-alkylidene anilines acting as electron-poor dienes. The primary Povarov cycloadduct is a tetrahydroquinoline that can be oxidized to the corresponding quinoline; therefore, it can be used to grant easy access to helical-shaped compounds (
Scheme 13) [
26].
Scheme 13. The Povarov approach to substituted azahelicenes.
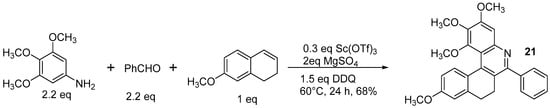
Actually, the reaction depicted in
Scheme 14 brings about the formation of the dihydroderivative
21, which cannot be further oxidized even in much harsher conditions (5 equiv. of DDQ, DMF, microwave 150 °C, 12 h).
Scheme 14. The formation of dihydroazahelicene 21 by the Povarov reaction.
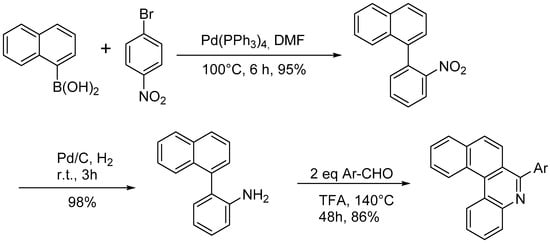
The Pictet-Spengler reaction was used to prepare a functionalized aza[4]helicene as depicted in
Scheme 15 [
27].
Scheme 15. Synthesis of aryl-substituted aza[4]helicenes by Pictet-Spengler reaction.
The reaction can be performed by both electron-deficient and electron-rich aldehydes, resulting in an aryl substituent that can bear groups such as –OCH3 or –NO2. In this paper, the optical properties of the aza[4]helicenes were studied by UV spectroscopy, finding absorption bands in CH2Cl2 with maxima at 274–307 nm, with the presence of electron-withdrawing groups resulting in absorption at longer wavelengths than the electron-donating groups; the positions of the substituents had no obvious effect on the maximum of the absorption peak. In the fluorescent spectra, the emission maxima for the helicene skeleton were observed at about 400 nm, and the substituent groups and their positions had no obvious effect on the emission, while the fluorescence intensities were weak. The methoxy group onto the aromatic ring can be demethylated with 98% yield by BBr3 in dichloromethane. This method could also be applied to azahelicenes with more than four rings.
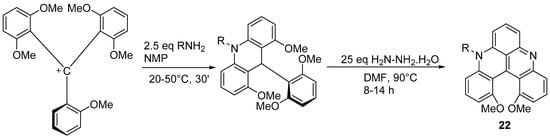
The configurationally stable diaza[4]helicene
22 is prepared starting from the tris(2,6-dimethoxyphenyl)methyl cation following the strategy indicated in
Scheme 16 [
28].
Scheme 16. Synthesis of a configurationally stable substituted diaza[4]helicene.
R can be alkyl, phenyl, benzyl, -NH2, -CH2CH2OH, or -CH2CH2 NH2. Yields range from 80 to 97%. This synthetic procedure is general, modular, and highly tolerant to functional groups. Single enantiomers of the [4]helicenes were obtained by chromatographic resolution, with a high racemization barrier. These helical quinacridines are effective dyes showing interesting absorption and emission properties, that can be modulated as a function of pH. Their fluorescence is weak and they show relatively modest changes in emissive properties upon protonation.
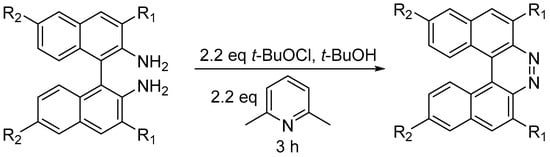
Substituted 7,8-diaza[5]helicenes were prepared by oxidative ring closure of 1,1′-binaphthalene-2,2′-diamines (BINAMs,
Scheme 17) [
29].
Scheme 17. Synthesis of substituted 7,8-diaza[5]helicenes.
t-BuOCl acts as bland oxidizing agent, while 2,6-lutidine serves as weak base to neutralize HCl generated in the process. The method is moderately tolerant to functional groups: R
1 can be H, CH
3, Br, Ph, and COOCH
3, while R
2 can be H, Br, or butyl. The reaction takes place at RT in 3 h with yields ranging between 44 and 91% [
29]. For these diazahelicenes, UV–VIS absorption spectra were recorded, observing a red shift over the whole region upon the introduction of methyl or phenyl groups in the 3,3′ positions, while on the opposite side the introduction of 6,6′-
n-Bu substituents caused a blue-shift of n–π * transitions (380–430 nm), while π–π * transitions (300–340 nm) were red-shifted. Cyclic voltammetry experiments were used to estimate the LUMO energies, which ranged from −2.92 to −3.13 eV.
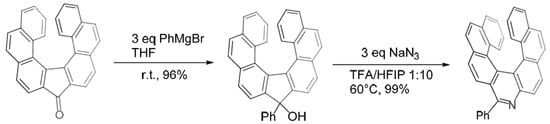
A very recent paper [
30] reported the synthesis of a substituted aza[7]helicene by ring expansion starting from a helical ketone, which was reacted with a carbanion to obtain a five-membered ring alcohol. This latter then undergoes ring expansion by reaction with NaN
3, with subsequent rearomatization/deprotonation to yield an aryl-substituted aza[7]helicene (
Scheme 18).
Scheme 18. Synthesis of aryl-substituted aza[7]helicenes by ring expansion.
A variety of mono- and poly-cyclic aryl moieties were attached to the aza[7]helicene with excellent yields through the use of an appropriate Grignard reagent; both electron-withdrawing and electron-donating substituents on the aryl ring were tolerated and even aliphatic chains could be introduced. The enantiomers were separated by chiral HPLC. Under the irradiation of UV light (365 nm), some of these compounds, notably those bearing Ph, n-Bu,p-OPh, and 7-(tert-butyl)pyren-2-yl, displayed a bright green luminescence. The quantum efficiency of the n-Bu-substituted derivative leaps to 18% in comparison to ∼7% for its aryl analogues. The longest absorption maximum in UV−VIS spectra is at ∼433 nm.