Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Calprotectin (CP), a heterodimer of S100A8 and S100A9 EF-hand calcium-binding proteins, is an integral part of the innate immune response. CP exploits the metal requirement of pathogens in the host immune response through the chelation of transition metals, starving pathogens of these nutrients. In addition, CP plays a role in the inflammatory response by acting as a ligand for cell surface receptors that signal through the NF-κB signaling pathway.
- calprotectin
- small molecule inhibitor
- inflammation
1. Introduction
Calprotectin is a unique heterodimer of two S100 EF-hand calcium binding proteins, S100A8 and S100A9. CP functions in the host innate immune response to infection by pathogens, exploiting their need for transition metals to grow and thrive [1]. CP inhibits the growth of pathogens by chelating various transition metals with high affinity, effectively starving them of these essential nutrients, a mechanism termed nutritional immunity. CP also plays a role in the host inflammatory response by acting as a ligand for pattern recognition receptors including the receptor for advanced glycation end products (RAGE), toll-like receptor 4 (TLR4), and cluster of differentiation 33 (CD33) [2,3]. These receptors are activated by a wide range of ligands and signal through a MAPK-dependent kinase cascade that activates the NF-κB transcription factor. The resultant expression of inflammatory cytokines and chemokines, and generation of reactive oxygen species serves to stimulate inflammation. In addition, this signaling cascade leads to increased expression of ligand and receptor, creating a pro-inflammatory environment, i.e., as CP is secreted from immune cells it generates a positive, pro-inflammatory feedback loop (Figure 1) [4,5].
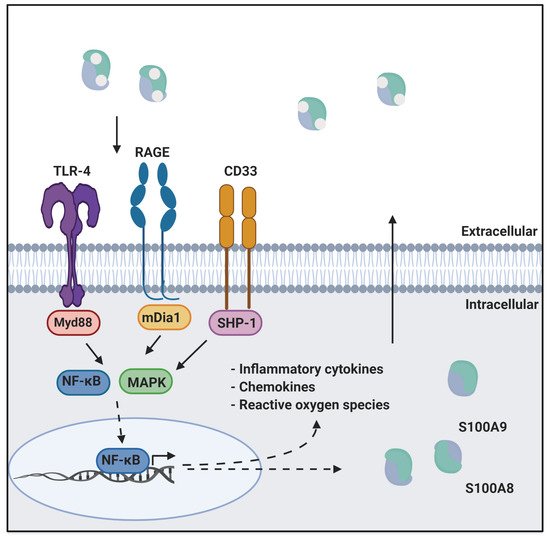
Figure 1. CP in innate immunity. CP serves as a ligand for inflammatory receptors such as RAGE and TLR4. Activation of these receptors through the MAPK dependent signaling cascade activates the NF-κB transcription factor. This results in the expression of cytokines, chemokines, and reactive oxygen species that drive inflammation. This figure was created using BioRender.com (last accessed on 10 February 2022).
Inflammation is an essential physiological response to microbial invasion as well as cell stress and injury. However, aberrant regulation leading to chronic inflammation arises in a number of pathological conditions, which can result in tissue damage and disease progression. Inflammation can be triggered externally during infection by microbial pathogen-associated molecular pattern molecules (PAMPs) or internally as a response to cellular damage by damage-associated molecular pattern molecules (DAMPs) such as CP and other S100 proteins. Increased expression of DAMPs has been shown to correlate with Alzheimer’s, cancers, and autoimmune diseases (reviewed in [6,7,8]). Mis-regulation of the expression of S100 protein DAMPS has been implicated in several diseases and a number of S100 proteins are in use in the clinic as biomarkers, including for a range of cancers [9].
One of the best studied examples of CP disease association is inflammatory bowel disease (IBD), a chronic inflammatory condition characterized by prolonged inflammation of the gastrointestinal tract and resulting tissue damage [10]. Inflammation in IBD has been shown to be caused by an accumulation of DAMPs as well as an increase in the expression of pattern recognition receptors such as RAGE [11,12,13]. In fact, overexpression of CP is used as a biomarker for the diagnosis of IBD and can serve as a predictor for disease prognosis [14]. IBD is treated with non-specific anti-inflammatory drugs targeting cytokines such as TNFα [15]. However, these drugs are not effective for all patients and can lose efficacy over time. CP represents a potential target for the treatment of IBD and in this vein, other disorders mediated through CP-driven inflammatory signaling.
2. CP as a Drug Target
Given the many connections of CP and other S100 proteins to chronic inflammation and specific diseases, there has been sustained interest in discovering, testing, and refining small molecule inhibitors for better understanding biochemical pathways and evaluating the potential therapeutic value of these molecules [59]. A variety of different approaches have been used to identify S100 protein inhibitors from direct small molecule screening to structure-based molecular design. In the following, we first provide an overview of progress organized by the various strategies used for small molecule discovery of S100 protein inhibitors, since these are all potential strategies that could be applied to CP. We then describe the current status of CP drug discovery and end with a brief overview of complementary progress to develop drugs that are targeted to the inflammatory receptors for which CP is a ligand.
2.1. Inhibitors of S100 Proteins
The first S100 protein inhibitors were found serendipitously in very broad, general searches to identify the target(s) of drugs whose mode of action was not firmly established at the time. One of the earliest studies involved the anti-allergy drugs amlexanox, cromolyn, and tranilast, which were known to inhibit IgE-meditated degranulation of mast cells [61]. Affinity chromatography experiments using bovine lung lysate identified a number of S100 proteins as binding partners. The interaction of specific S100 proteins with these molecules was then investigated. Binding of tranilast to S100A12 was shown to block the interaction with the RAGE V domain [37]. Similarly, binding of cromolyn to S100P was shown to block interaction with RAGE [62]. Analogs of cromolyn have been developed that show enhanced binding to S100P, which was proposed to minimize its off-target effects [63]. However, this hypothesis has yet to be tested. Although these molecules show chemotherapeutic effects in cells, they are not specific for any one S100 protein and have been shown to interact with a number of kinases as well [64,65]. Similarly, the phenothiazine class of antipsychotic drugs were shown using affinity chromatography to bind to S100B and S100A1 as well as other EF-hand calcium-binding proteins [64]. Although these molecules bind a number of EF-hand proteins, structural studies have revealed that the binding modes for each protein are quite different [66,67,68]. Efforts to improve specificity of these molecules are ongoing [64,69,70].
A number of S100 protein drug development efforts have utilized high throughput screening (HTS) combined with structure-based molecular design. A fluorescence polarization competition assay (FPCA) was developed for HTS of S100B to selectively screen small libraries of compounds that can bind Ca2+-bound S100B and inhibit its interaction with a p53 peptide in vitro [68]. The assay was based on the peptide termed TRTK-12, which is known to bind at the S100B target interface. The peptide was labeled with a fluorophore and changes in fluorescence anisotropy were used to monitor release from S100B when it was outcompeted by a ‘hit’. In another campaign, an inhibitor screen of FDA approved drugs was carried out for S100A4 using a fluorescent biosensor and protein conjugate, Mero-S100A4 [71]. The Mero biosensor is linked covalently to S100A4 via Cys81 and binds along the target binding interface of S100A4 with two molecules of Mero bound per S100A4 dimer. This screen identified a number of molecules that bind S100A4, including phenothiazines. An X-ray crystal structure of phenothiazine trifluoperazine (TFP) bound to S100A4 revealed two TFP molecules bound in the hydrophobic cleft of S100A4 with four molecules bound per dimer [72]. This 4:1 ratio was sufficient to induce oligomerization of S100A4 to a pentamer. Interestingly, inhibition of the S100A4 function was observed only when the concentration of TFP was high enough to induce oligomerization [72].
Virtual screening has been used as a starting point for some S100 protein drug discovery programs. After screening the large number of potential hit molecules in silico, hits are verified by biophysical methods such as NMR. Verification of hits by NMR for S100B enabled prioritization of those interacting at the target binding interface. The structure of S100B in complex with one such hit, SEN205A, was then determined by X-ray crystallography [73]. In another study, inhibitors of S100A10 were discovered using virtual screening and subsequent analysis by a competitive fluorescence binding assay [74]. After the optimization of hits based on structural data, analogs of 4-aroyl-3-hydroxy-5-phenyl-1H-pyrrol-2(5H)-one and substituted 1,2,4-triazoles were synthesized and then shown to inhibit the interaction of S100A10 with an annexin A2 peptide [74].
After screening to find hit molecules, structure-guided elaboration is greatly facilitated by knowledge of target binding sites, which have been analyzed for their unique chemical and structural characteristics [75]. Moreover, there are numerous structures of S100 proteins bound to peptide fragments of their targets. Among these, the interaction of S100B and its target p53 is one of the most well characterized (reviewed in [76]). This interaction is calcium-dependent; S100B, like other S100 proteins, undergoes a conformational change upon calcium-binding that exposes a hydrophobic patch that is essential for binding to p53. A virtual screen using high resolution crystal structures of S100B and the S100B–p53 complex was used to screen a library of compounds to predict inhibitors [72]. Seven of the virtual hit molecules were found to inhibit the S100B interaction with a p53 peptide, one of which was the FDA approved drug pentamidine. Chemical elaboration of pentamidine using a structure-guided approach resulted in tighter binding to the protein [69].
The target-binding site of S100B has also been characterized by structures of complexes with small molecule inhibitors (Figure 3). Interestingly, these bind in a variety of different modes. The hydrophobic patch of S100B has been subcategorized into three distinct pockets, termed Site 1, Site 2, and Site 3 [69]. Site 1 is the p53 target binding site where p53 fragments interact with Helices 3 and 4, as well as a peptide derived from RAGE (Figure 3B,C). The above noted small molecule SEN205A binds in this Site 1 region, inhibiting by directly blocking the p53 binding site. Surprisingly, the majority of S100B inhibitors such as pentamidine and heptamidine bind at Site 2 and Site 3, interacting with Helix 4 and the loop region on Helix 2 (Figure 3D,E). As these inhibitors are able to block p53 binding without binding directly in the target binding site, they are presumably inducing allosteric structural effects that disrupt target binding. A structure-based approach has been used to further optimize these compounds, and a number of these S100B-inhibitor complexes have been structurally characterized [70]. All ‘hit’ compounds were tested for inhibition in the presence and absence of calcium to make sure that they are inhibiting the relevant form of the protein [70]. Binding to S100A1 was also investigated to begin to test the specificity of these molecules among the S100 proteins.
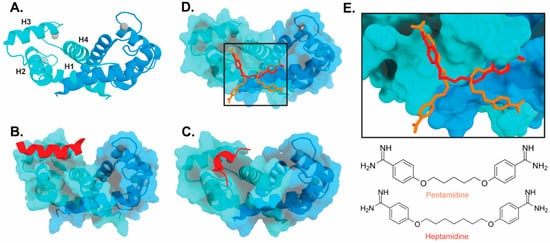
Figure 3. Binding of ligands to S100B: (A) Ribbon structure of calcium-bound S100B dimer (PDB 3D10) individual subunits are marked in blue and cyan with calcium represented as gray spheres. The four helices of one S100B subunit are labeled H1–H4. (B) S100B bound to p53-derived peptide (PDB 1DT7). (C) S100B bound to RAGE-derived peptide W61 (4XYN). (D) S100B bound to two molecules of pentamidine and one molecule of heptamidine (4FQO). (E) Zoomed in view of panel D binding site. Chemical structures are shown below.
2.2. Inhibitors of Calprotectin
The large majority of drug discovery efforts for CP derive from an ongoing program at the company Active Biotech (Lund, Sweden). This effort originated with a broad screen to identify novel anti-inflammatory molecules, which turned up a number of quinoline-3-carboxamides (Q compounds) with interesting anti-cancer and immunomodulatory properties [77]. The focus of the program was initially on roquinimex, but it and other analogs were found to elicit pro-inflammatory effects [77]. In order to reduce the pro-inflammatory properties of these molecules, a number of second-generation Q-compounds were synthesized and tested for anti-inflammatory activity in animal models for multiple sclerosis. This resulted in a number of newer lead molecules, including laquinimod (ABR-215062), tasquinimod (ABR-215050), and paquinimod (ABR-215757) [78] (Figure 4). These three molecules are currently in Phase 2 and 3 clinical trials for the treatment of various cancers and inflammatory disorders such as multiple myeloma, prostate cancer, and uveitis. Five years ago, Active Biotech released for study ABR-238901, which is purported to be an inhibitor of both S100A9 homodimer and CP. This molecule has been used in multiple studies in cells and mouse models of inflammatory diseases, revealing that treatment with ABR-238901 dampens inflammation [79,80,81]. However, no data have been reported that directly confirm this molecule binds with significant affinity to S100A9 homodimers or CP, or that in cells, this molecule binds only to S100 proteins.
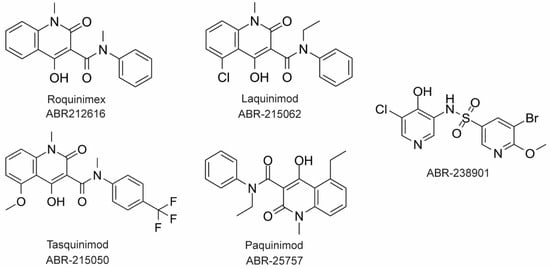
Figure 4. Chemical structures of Active Biotech CP inhibitors reported to date.
The potential therapeutic value of these S100A9-binding Q compounds has been investigated in several models. It has been proposed that Q-compounds inhibit tumor growth by blocking S100A9 activation of TLR4-mediated inflammation [82]. In support of this hypothesis, treatment with tasqinimod has been shown to inhibit tumor growth in a mouse model of prostate cancer in a manner that mimics the phenotype seen in S100A9 deficient mice [83]. In addition, S100A9 is known to be secreted from myeloid-derived suppressor cells (MDSC) and serves as a chemoattractant that promotes MDSC recruitment and tumor signaling pathways. Therefore, the anti-carcinogenic effects of S100A9-binding compounds in this context appear to arise from inhibition of S100A9-dependent recruitment of MDSCs [82].
In an investigation into the mechanism of action of Q-compounds the authors concluded that they meditate inflammation by directly binding to S100A9 [84]. In that study, biophysical analysis in vitro showed that Q-compounds interact specifically with S100A9 homodimers and not CP or S100A8 homodimers. In addition, binding of Q-compounds to S100A9 assayed by surface plasmon resonance was reported to block binding to RAGE and TLR4. Despite evidence to the contrary in the literature, the authors proposed that only the S100A9 homodimer and not CP bind to RAGE and TLR4 [84]; further investigation will be required to further test the generality of this hypothesis.
2.3. Receptor-Targeted Drugs
Because of its role in inflammation, there has been significant interest in RAGE as a therapeutic target [85]. RAGE-binding peptides have been designed from the structure of one of its ligands, S100P [86]. In addition, the small molecules including Azeliragon and FPS-ZM1 [87] have been shown to inhibit the activity of RAGE [88]. In vitro biochemical data show that these molecules are able to inhibit RAGE activation by different binding partners [87,89]. However, there are no structural data that would define the mechanism of inhibition for any of these molecules. Despite the intense interest and existence of RAGE inhibitors molecules, with some in clinical trials, there are no FDA approved drugs for RAGE [90].
TLR4-targeted drugs are also of intense interest [91] in the context of a variety of autoimmune diseases [92], infection-induced sepsis [93], and inflammatory bowel disease [94], all associated with S100 proteins. A number of small molecule inhibitors have been developed to inhibit TLR-mediated inflammation pathways. TAK-242 is a small molecule inhibitor that has been shown to bind to TLR-4 and block interactions with its adaptor molecules [95]. Although this inhibitor was shown to be specific for TLR-4, it did not make it past Phase 3 clinical trials as it was not able to significantly suppress serum cytokine levels in septic patients compared to controls. Other small molecule inhibitors such as Corilagin [96] and Artesunate [97] have been shown to block LPS-induced inflammation in a TLR-4 dependent manner. However, here again, the absence of structural information has limited the understanding of the mechanism of action of these drugs.
CD33 is also an attractive therapeutic target for acute myeloid leukemia (AML) and neurodegenerative disorders. There have been clinical trials on drug–antibody conjugates that target CD33 [98]. In addition, targeting CD33 with sialic acid mimetics has been successful in inhibiting CD33 in vitro [99]. There are, however, no reports of drug-like molecules that bind CD33.
This entry is adapted from the peer-reviewed paper 10.3390/biom12040519
This entry is offline, you can click here to edit this entry!