Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Berbamine is a natural, potent, pharmacologically active biomolecule isolated from Berberis amurensis. Berbamine has been shown to modulate different oncogenic cell-signaling pathways in different cancers.
- cancers
- berbamine
1. Targeting of Ca2+/Calmodulin-Dependent Protein Kinase II (CAMKII) by Berbamine in Different Cancers
Targeting c-Myc with small-molecule inhibitors remains challenging. c-Myc protein stability can be controlled by phosphorylation at two different sites with opposing functions. Phosphorylation at 62nd serine residue stabilized c-Myc, whereas phosphorylation at 58th threonine residue promoted the degradation of c-Myc. Ca2+/calmodulin-dependent protein kinase II (CAMKII), a multifunctional serine/threonine kinase, has been shown to stabilize the oncogenic c-Myc level [1]. Expectedly, levels of phosphorylated-c-Myc (62nd serine residue) and total c-Myc were noted to be reduced in CAMKIIγ knockdown cells, whereas levels of phosphorylated-c-Myc (62nd serine residue) and total c-Myc were found to be significantly enhanced in CAMKIIγ overexpressing T cell lymphoma cells [1].
An orally administered, bioactive small molecule analog of berbamine, tosyl chloride-berbamine (TCB), considerably reduced phosphorylated levels of CaMKIIγ [2]. TCB induced a regression of leukemia growth in an orthotopic B-ALL model using NSG (NOD/SCID/IL2Rγ-/-) mice injected with CaMKIIγ/Myc-expressing leukemia cells [2].
Berbamine had the ability to bind to the ATP-binding pocket of CaMKIIγ, inhibiting its phosphorylation and inducing apoptosis in leukemia stem cells [3]. 4-Chlorobenzoyl berbamine (CBBM), a Berbamine derivative, effectively enhanced the proteasome-dependent degradation of c-Myc in OCI-Ly3 cells [4].
Berbamine has been reported to block VEGF- and BDNF-regulated angiogenesis, mainly through the inactivation of VEGFR2- and TrkB-mediated transduction cascades [5]. Berbamine considerably reduced VEGF-dependent phosphorylation of VEGFR2, as well as that of TrkB by BDNF in HUVECs, resulting in the deactivation of downstream effectors, such as PKB/AKT, NF-κB and ERK1/2. Berbamine efficiently reduced BDNF and VEGF-mediated CaMKIIγ phosphorylation. Berbamine significantly suppressed tumor growth in chorioallantoic membrane tumor models implanted with U87MG cells [5].
Berbamine and one of its derivatives, BBD24, strongly inhibited CAMKII phosphorylation in Huh7 cells [6]. Overall, these studies helped us develop a sharper understanding of the instrumental role of the CAMKII/c-Myc-signaling axis in carcinogenesis.
2. Regulation of Autophagy by Berbamine
Berbamine is a natural molecule from traditional Chinese medicine that is useful for the treatment of patients with inflammation and cancers such as leukaemia, lung, liver and breast cancer. Berbamine is administered to patients with leukopenia caused by conventional chemotherapy and/or radiotherapy. Several reports indicate that berbamine has a role in the modulation of deregulated pathways in cancers. Berbamine causes caspase-3-dependent apoptosis in leukaemia cells through surviving pathway activation [7]. Moreover, berbamine inhibits the cell growth and motility of highly metastatic breast cancer and lung cancer cells [8][9].
Recently, berbamine has been considered a novel autophagy inhibitor in breast and colon cancer cells. Autophagy is an essential catabolic process involved in many pathological conditions, including cancer, that can protect cells and organisms from stressors. The role of autophagy in cancer remains uncertain and has been reported as a pro- and anti-tumorigenic system [10]. In pre-malignant lesions, autophagy activation might prevent cancer development [11]. Conversely, in advanced cancers, autophagy induction can stimulate carcinogenesis, such as in melanoma, colorectal, pancreas and renal cancers, or suppress it, as in breast cancer [12][13][14][15][16][17]. In 1988, Ohsumi described, for the first time, autophagy mechanisms as a lysosomal degradation and cellular recycling pathway, evolutionarily conserved, that allows protein aggregates and damaged organelles to be eliminated through lysosomal degradation, thus maintaining cellular homeostasis [18]. Autophagy can be divided into three groups: macroautophagy, microautophagy and chaperon-mediated autophagy. Macroautophagy (refer to macroautophagy as autophagy) is the best known of the three pathways. Autophagy mediates the sequestration and delivery of cytoplasmic material to lysosome for degradation. This process induces the phagophore formation caused by the extension of an isolated membrane, which fuses to convey cytoplasmic components into an autophagic double membrane vacuole, the autophagosomes; the organelles fused with lysosomes become autolysosomes, which degrade the materials contained within it. The autophagy process is modulated at transcriptional and post-translational levels, and the genes involved in autophagy are regulated by ATF4 at the transcriptional level. Cellular stresses induce MIT/TFE transcription factors and other inhibitors of mTOR, a negative regulator of MIT/TFE. In the autophagy process, the fusion of membranes is usually realized by soluble SNARE complexes (N-ethylmaleimide-sensitive factor attachment protein receptor). Recently, it has been described that the SNARE syntaxin 17 (STX17) contained in autophagosomes interacts with the cytoplasmatic SNARE SNAP29 and SNARE VAMP8 of the lysosomes, and these proteins cooperate in autophagosome-lysosome fusion.
Fu et al. reported that berbamine causes the upregulation of BNIP3, inhibiting SNARE-mediated autophagy-lysosome fusion in breast cancer cells. Under hypoxia, BNIP3, a protein with homology to BCL2 in the BH3 domain, drives mitophagy in many different cell types. BNIP3 may play a role in autophagosome-lysosome fusion regulation. Berbamine induces the upregulation of BNIP3, which binds SNAP29 and inhibits the interaction between SNAP29 and VAMP8, which, in turn, causes a blockade of autophagosome-lysosome fusion and autophagosome increase (Figure 1) [19]. This study suggests that berbamine could be considered a potential autophagy inhibitor, which could be used in combination with chemotherapy in cancer management. Autophagic cell death can also be induced by RAS/RAF/MEK/ERK pathway activation [20]. Mou et al. reported that berbamine can exert anticancer effects on human colon cancer cells, inducing autophagy and apoptosis and inhibiting cell migration via the MEK/ERK pathway. Berbamine induces autophagic vesicles in colon cancer cells and an increase in Beclin-1, LC3B-II, ATG-5 and 12 expression [21].
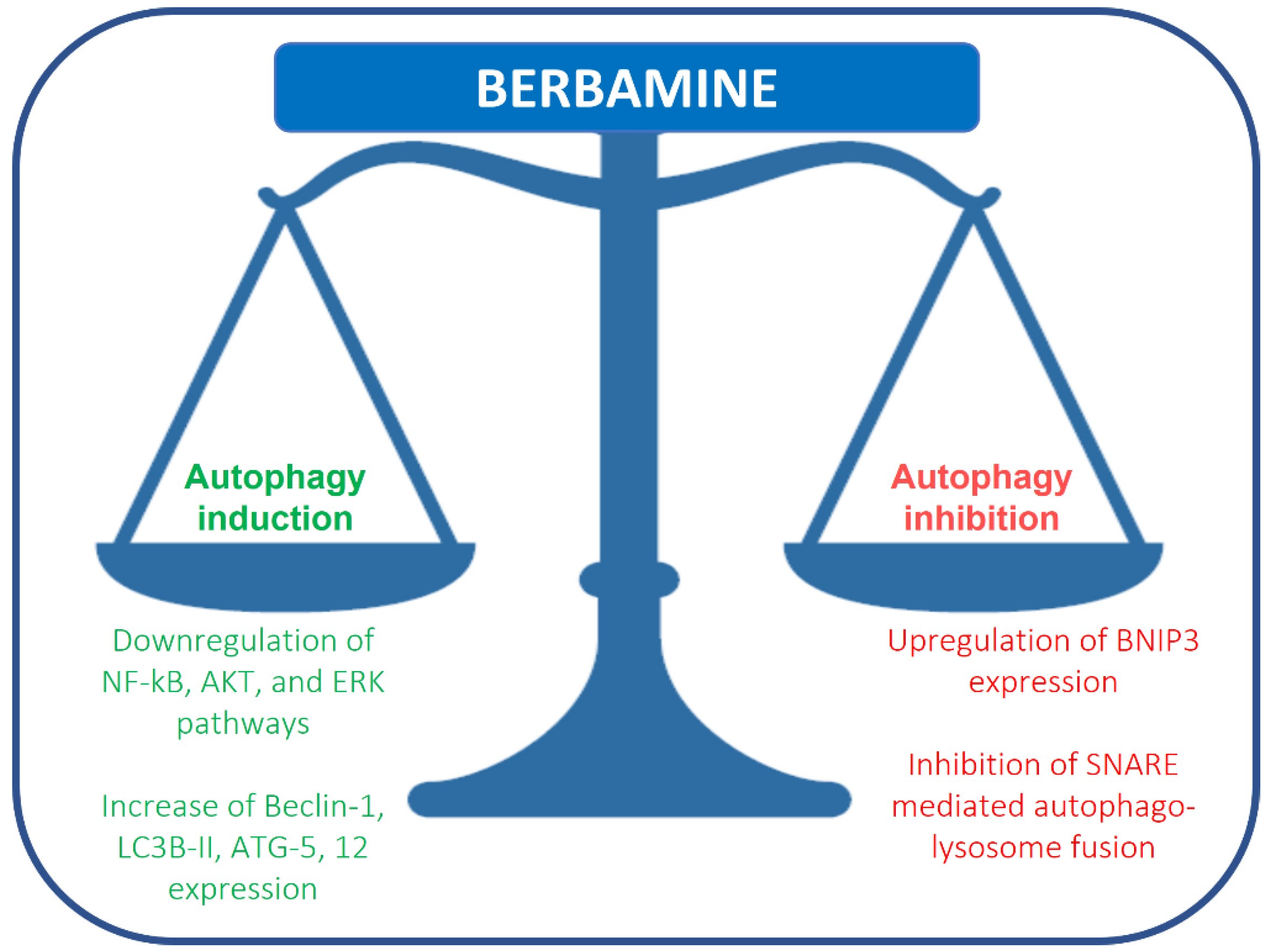
Figure 1. Regulation of autophagy by berbamine.
Preliminary studies on berbamine indicate its role in cancer treatment. It is efficient because other natural compounds could be limited by reduced water solubility, low absorption rate in the bowels and rapid metabolism. The role of berbamine in autophagy is still unclear. Further studies are needed to elucidate the effects of this natural compound on autophagy regulation.
3. Targeting of Protein Networks by Berbamine for Cancer Chemoprevention
Berbamine efficiently suppressed the migratory and invasive capacity of highly metastatic breast MDA-MB-231 cancer cells via the inhibition of pro-MMP2/MMP9 activation. It also reduced the phosphorylated levels of AKT and c-Met in MDA-MB-231 cancer cells [9].
Aspirin dose-dependently induced the phosphorylation of CREB and ATF1 [22]. Importantly, the treatment of HepG2 cells with AICAR (an AMPK agonist) also resulted in the phosphorylation of CREB and ATF1. However, knockdown of AMPKα1 abolished the phosphorylation of CREB/ATF1 by aspirin in Hep3B and HepG2 cells. PKA (Protein kinase A) is present downstream to AMPK and mediates the phosphorylation of CREB/ATF1 by aspirin. Accordingly, PKA inhibitors impaired the phosphorylation of CREB/ATF1 by aspirin in HepG2 and Hep3B cells. Soluble adenylyl cyclase (sAC) has an important role in cAMP synthesis. Aspirin did not induce phosphorylation of CREB/ATF1 in sAC knockdown cells. Aspirin caused marked reduction in the levels of CPS1 (carbamoyl phosphate synthetase I) in HepG2 and Hep3B cells. Similarly, AICAR (an AMPK agonist) inhibited CPS1 expression in HepG2 and Hep3B cells. AMPK knockdown abrogated the downregulation of CPS1 and upregulation of sAC by aspirin. CREB/ATF1 knockdown sensitized Hep3B and HepG2 cells to aspirin. CREB/ATF1 activation antagonized the anti-cancer effects of aspirin, and pharmacological targeting of CREB/ATF1 significantly enhanced the efficacy of aspirin against HCC cancer cells. Berbamine inhibited CREB/ATF1 phosphorylation and sensitized HCC cells to aspirin. Protein phosphatase-2A (PP2A) induced dephosphorylation of its substrates. However, CIP2A (a cellular inhibitor Of PP2A) negatively regulated PP2A. Berbamine reduced CIP2A levels in HCC cells and promoted PP2A-mediated abrogation of aspirin-induced phosphorylation of CREB/ATF1 [22].
Berbamine concentration-dependently inhibited the migratory and invasive potential of SMMC-7721 cells and increased expression of Cx32 in SMMC-7721 cells. However, after silencing Cx32, berbamine failed to inhibit cell invasion and metastasis [23].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23052758
This entry is offline, you can click here to edit this entry!