1. Introduction
Neurodegenerative diseases are age-dependent disorders characterized by the progressive degeneration of neural cells, affecting nearly 57 million people worldwide and 10 million new cases every year
[1]. These diseases impose substantial medical and public health burdens
[1]. They are increasingly prevalent and incidental, mainly due to an increase in life expectancy, and are expected to increase dramatically in the near future
[1]. The etiological factors of these diseases are poorly understood and result from genetic and environmental factors
[2][3]. Mostly in high-income countries, diagnosis can combine clinical examination, including neuropsychological testing, brain imaging (e.g., 18F-fluorodeoxyglucose positron emission tomography (PET), magnetic resonance imaging, and amyloid or tau PET)
[4][5], and cerebrospinal fluid (CSF) biomarkers in the case of Alzheimer’s disease (AD)
[6]. Particularly, neuroimaging associated with CSF biomarkers was introduced to increase the diagnostic accuracy of AD, especially at the early stage and in the case of atypical clinical presentation
[7]. Currently performed on CSF, most clinical laboratory tests combine elevated levels of total and phosphorylated tau proteins (e.g., pT181 and pT217) and reduced soluble amyloid-beta peptide (Aβ)42 levels or Aβ42/40 ratios allow distinguishing AD patients from non-AD individuals with a mean accuracy of 85–89%
[8][9]. Such biomarkers are currently unavailable for non-AD neurodegenerative diseases. Besides, no validated blood-, saliva- or urine-based biomarkers and no specific therapy is currently available for clinical use in neurodegenerative diseases
[4][5]. According to the anatomical tropism of the neurodegenerative processes, i.e., the specifically affected brain tissue areas, some of these diseases start with cognitive or behavioral impairments (dementia), while others first begin with movement disorders, sensory-motor deficits, or epilepsy
[10][11][12][13]. The broad spectrum of primary neurodegenerative disorders mainly includes AD, frontotemporal lobar degeneration (FTLD), dementia with Lewy bodies (DLB), Parkinson’s disease (PD), multiple system atrophy (MSA), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Creutzfeldt–Jakob disease (CJD), amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD)
[14]. There is a substantial overlap in the clinical symptoms of these diseases, which complicates an effective and accurate diagnosis, particularly in the early stages
[4][15]. Besides, co-occurring pathologies are frequent and further blur the clinical phenotype boundaries, representing a challenge for identifying specific biomarkers
[16].
While abnormal protein aggregates typically define neurodegenerative diseases, it is unclear whether these abnormalities are driving the disease or are themselves consequences of other underlying processes
[17]. Several fundamental processes are associated with progressive neuronal dysfunction and death, including abnormal protein dynamics, proteotoxic stress, dysfunctions in the ubiquitin-proteasome and autophagosomal/lysosomal systems, oxidative stress, programmed cell death, and neuroinflammation
[18]. Abnormal protein aggregates can take several histopathological forms in the brain tissue: neurofibrillary tangles, Pick’s bodies, tufted astrocytes, Lewy bodies, amyloid plaques, among others
[18]. For instance, AD is a dual proteinopathy characterized by accumulating tau aggregates in neurofibrillary tangles and extracellular aggregates of Aβ plaques
[19][20]. The aggregation of α-synuclein into Lewy bodies and neurites is characteristic of synucleinopathies, including PD and DLB, while glial cytoplasmic inclusions of α-synuclein in oligodendrocytes can occur in MSA
[21][22]. The accumulation of tau 4R isoforms, i.e., isoforms that contain four carboxy-terminal repeat domains, in neuronal and glial cells is the main characteristic of PSP and CBD. The transactive response DNA binding protein-43 (TDP-43) is the main component of intracellular ubiquitin inclusion bodies in pathological deposits in ALS and FTLD
[12][18]. Several challenges, including the complexity of protein aggregation, misfolding, pathology propagation processes, and the immune response, need to be explored for successful translational research on neurodegenerative disorders.
The discovery of biomarkers in neurodegenerative diseases remains an important challenge in modern medicine
[17]. An ideal biomarker should reflect pathological changes in the brain with accurate performance, differentiating forms of neurodegenerative diseases
[23]. Currently, neurodegenerative disease research for biomarker discovery is focused on: (1) early disease biomarkers, present in both brain tissue and biological fluids (e.g., CSF and blood)
[8][9][23][24][25]; (2) detection of molecular signatures, i.e., the combination of proteins or proteoforms (more details about proteoforms can be found elsewhere
[26]), that correlate with the neuropathological processes or disease status (e.g., rate of progression, treatment response)
[23][27][28][29]; and, (3) surrogate biomarkers for developing disease-modifying therapies
[30][31][32][33][34].
Mass spectrometry (MS) is characterized by high detection sensitivity, specificity, and multiplexing capability for biomarker discovery and validation
[35]. MS-based proteomics has considerably extended our knowledge about the occurrence and dynamics of protein post-translational modifications (PTMs). Profiling the main protein players of neurodegeneration in the brain using MS can provide new insights into the role of aberrant PTM patterns in the pathological process and identify signatures with potential diagnostic or therapeutic relevance.
2. The Next Step in Neuroproteomics
Although it has enjoyed remarkable recent success stories, proteomics still faces substantial technical challenges. Proteomics data processing and analysis is a multistep process that requires a lot of expertise. Multiple stages are necessary for the consistent analysis of LC-MS and LC-MS/MS data. Adequate sample preparation, reducing complexity, and enriching lower abundance components while depleting the most abundant can help solve the analysis challenges. Sample enrichment using immunoprecipitation of the target protein, fractionation, or PTM-specific enrichment methods (chemical and biochemical) are now pretty well established
[36]. Various offline fractionation methods have been employed to enhance the depth of the proteome by improving the detection of low abundance peptides before LC-MS/MS analysis. Methods including two-dimensional gel electrophoresis
[37], strong cation exchange (SCX), electrostatic repulsion-hydrophilic interaction chromatography (ERLIC)
[30], and high-pH reversed-phase chromatography
[38] are used to increase peptide identification by separating peptides in an orthogonal dimension. Unfortunately, sample processing is still the main bottleneck for many extensive proteomics studies because it is a complex multiple-step process. There is no standard best approach for sample processing, and optimizing this step is detrimental to the ability to study the proteoform(s) of interest effectively.
Additionally, state-of-the-art mass spectrometry instrumentation and extensive automatized high-confident data processing and analysis are required. Label-free quantification, super-SILAC, and chemical labels can be employed for large-scale quantitative discovery. Modified peptides that may serve as biomarkers can be validated with larger cohorts using targeted MS methods such as MRM or PRM. Finally, with MS instrumentation improving, the bottleneck shifts towards confirming PTM sites and validating their function using new experimental and computational strategies. The exploitation of this technology resulted in significantly enhanced protein sequence coverage, the discovery of undescribed modifications, and the parallel analysis of different types of modification sites. These studies also generally characterized only a limited number of PTM types, with the most robust emphasis on phosphorylation and little focus on less common PTMs such as acetylation or glycosylation.
Furthermore, most MS-based experiments employed DDA, a data collection mode that relies on the “detectability” of the peptide species of interest, biasing the analysis towards peptides of the highest intensity. This particularly handicaps the identification of PTMs, as the modified species can be present in very low stoichiometries and/or exhibit lower detectability compared to their unmodified counterpart
[39]. In addition, studies have now tried to understand the impact of PTM crosstalk
[40]. Notably, the combination of PTMs, rather than individual PTMs, define protein function and are involved in the pathological mechanisms underlying neurodegenerative diseases. However, the current limitations in large-scale PTM combinatorial analyses render developing adequate clinical biomarker assays that target peptides with different modifications extremely challenging. This might be facilitated in the future with the continuous development of next-generation mass spectrometry workflows, i.e., top-down or native MS
[41][42].
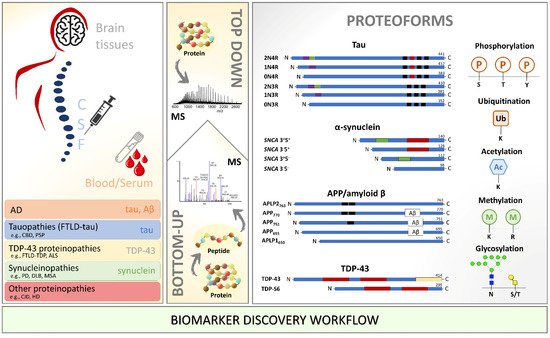
There is optimistic thinking about what the next few decades in the neuroscience field will bring. With the neurodegenerative cases increasing as the global population ages, a significant effort will have to be made to improve the complete integration of robust but sensitive MS-based proteomics approaches into biomarker discovery facilities (
Figure 1) and, ultimately, into clinical settings. First, however, it is crucial to understand (1) how PTMs regulate the cellular events; (2) how to rank by importance the several PTMs occurring on a single protein; and (3) what are the switch on-and-off responses and crosstalks of PTMs to pathological events, considering a particular pathological tissue or biofluid in a certain disease stage. The aim is to identify unique pathological signatures and biomarkers that can aid in understanding and monitoring the different processes involved in disease development and progression. For this, it is vital to limit inconsistencies across clinical studies by studying well-classified patient material and large patient cohorts and standardizing methodologies and protocols. Therefore, the field must now focus on large-scale multicentric MS-based translational studies integrating molecular signatures with pathological findings to diagnose and stratify neurodegenerative diseases. Integrative proteomic and genomic/transcriptomic analyses (in neuroproteogenomics settings) hold great promise for understanding the role of the PTMome in neurodegenerative diseases. For example, a multi-omics data fusion from 177 studies and more than one million patients with AD, PD, HD, and ALS has recently shed light on different biological processes between these pathologies
[43]. However, PTMome information was not considered in this integrative study.
Figure 1. Biomarker discovery workflow using mass spectrometry for identification and characterization of proteoforms in the different biological milieu of patients with neurodegenerative diseases. Amyloid β: amyloid-beta; APP: amyloid-beta precursor; ALS: amyotrophic lateral sclerosis; CBD: corticobasal degeneration; CJD: Creutzfeldt–Jakob disease; CSF: cerebrospinal fluid; DLB: dementia with Lewy bodies; FTLD: frontotemporal lobar degeneration; HD: Huntington’s disease; MS: mass spectrometry; MSA: multiple system atrophy; PD: Parkinson’s disease; PSP: progressive supranuclear palsy; SNCA: synuclein alpha; TDP-43: TAR DNA-binding protein 43.
Interestingly, by including metabolomics approaches, it is possible to understand the PTM relative changes by monitoring the precursors of protein PTMs (e.g., acetyl-CoA for acetylation, ATP for phosphorylation, and S-adenosylmethionine for methylation)
[44]. Therefore, analysis of PTMs in multi-omics studies will almost certainly reveal novel and even unexpected molecular signatures. This will allow for a better understanding of the molecular pathways underlying the development of neurodegenerative diseases, thereby facilitating the decision-making process and precision medicine settings. Although these integrative approaches are promising, there is still a long way to go to routinely incorporate omics data into clinical decisions for personalized interventions.
3. Concluding Remarks
High-throughput proteomics and PTM-omics studies using MS have been used in recent years to investigate neurodegenerative diseases. Great potential is underlying specific pathological PTM-signatures for clinical applications as biomarkers in neurodegenerative diseases. This is the case of diverse modified peptides from the major players in neurodegeneration processes such as tau, α-synuclein, APP/amyloid-beta, or TDP-43. Tau protein remains the most well-studied protein using MS. Several phosphorylated, acetylated, ubiquitinated, methylated, or glycosylated tau peptides are accumulated in the brain of AD and CBD or associated with a higher AD Braak stage. In addition, phosphorylated and C-terminal truncated forms of tau can be detected in CSF and serum in AD. For α-synuclein, studies focus more on phosphorylated, ubiquitinated, and truncated proteoforms that could be overexpressed specifically in tissues, CSF, and serum from DLB or PD patients. To the best of our knowledge, TDP-43 truncated forms were only studied in ALS brain tissues, and amyloid-beta was only analyzed in AD CSF. We believe that major breakthroughs can occur in detecting and characterizing PTMs less studied in the field using HR/MS. Due to the lack of data on non-AD neurodegenerative diseases, we also envisage that these will undoubtedly be the focus of neuroproteomics studies.
Adapted from Azevedo, R.; Jacquemin, C.; Villain, N.; Fenaille, F.; Lamari, F.; Becher, F. Mass Spectrometry for Neurobiomarker Discovery: The Relevance of Post-Translational Modifications. Cells
2022, 11, 1279.
https://doi.org/10.3390/cells11081279