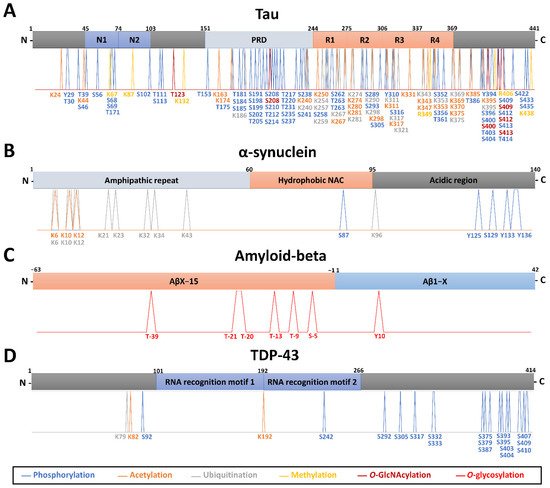
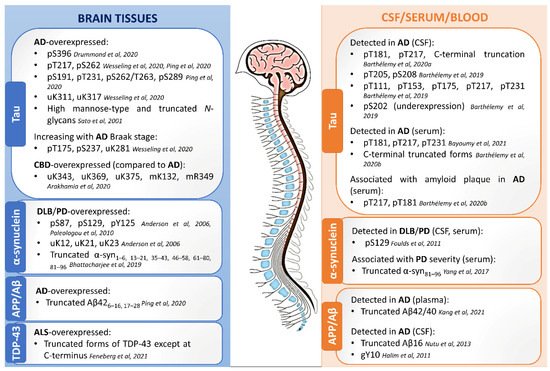
Profiling the PTM-modified proteins from the neuropathological hallmark aggregates, including tau, α-synuclein, amyloid-beta, and TDP-43, has been performed in different biological milieus using MS. A summary of the main findings of PTM localization sites identified by MS is found in Figure 1. An overview of the relevant PTMs identified by MS with potential for diagnosis or detection of disease progression in neurodegenerative diseases is illustrated in Figure 2.
3.2.1. Tau Protein
Phosphorylation of the microtubule-associated protein tau is one of the most important PTM for axonal stabilization and regulation [
99]. Still, abnormally phosphorylated tau (P-tau, phosphorylated at non-physiological sites) appears to lead to neurofibrillary tangles (NFTs) that no longer stabilize microtubules [
100]. Numerous research, including large-scale studies [
31], have consistently reported that tau levels (total tau and hyperphosphorylated tau) are prominently increased in the AD brain tissues [
67,
101], CSF [
102], and plasma [
85]. Total tau is linked to the severity of neurodegeneration, whereas phosphorylated tau reflects specific AD pathological changes in the neurofibrillary system [
103]. Tau hyperphosphorylation seems to be required but is insufficient to induce tau aggregation; other less investigated tau PTMs are certainly involved [
104,
105]. For example, several other PTMs, including ubiquitination and methylation, influence tau filament structure by contributing to the structural diversity of tauopathy strains [
86] and may play an important role in tau localization and protein–protein interactions [
106]. Analysis and quantification of tau PTMs in the brain and biofluids by MS is mainly focused on AD and CBD and is still missing for the other neurodegenerative diseases.
Brain tissues. Brain soluble tau phosphorylation sites are mainly localized at the C-terminus, at proline-rich mid-domain, and a cluster on the N-terminal projection domain [
67]. Tau’s longest isoform (2N4R, 441 aa) has 85 potential phosphorylation sites, and almost 20 residues were found to undergo phosphorylation in the healthy brain [
107]. In brain tissues from seven patients with advanced sporadic AD, LC-MS identified 542 proteins in NFTs and tau in all seven cases [
34]. Tau was phosphorylated on 23 different residues, and the most abundant tau phosphosite was pS396 [
34]. Other studies published before the year 2000 report various tau phosphorylation sites in AD after enrichment with monoclonal antibodies against tau and MS analysis [
107,
108,
109]. Recently, Barthélemy et al. [
67] increased the number of known tau phosphorylated sites to 29, identified pS404 as the most abundant species in the brain, and 12 of these phosphopeptides were common to CSF in AD by an in-depth targeted MS (PRM) analysis, independently of tau concentration. Phosphorylation of some particular sites was exacerbated (or specifically detected in AD) compared to controls, supporting the hypothesis that tau phosphorylation could be a physiological process amplified by AD pathology [
67]. Tau phosphopeptides pS191, pT217, pT231, pS262, pS262/T263, and pS289 showed a significant increase in AD compared with control samples [
31]. Using an exploratory IP-MS approach, tau phosphorylation on brain soluble fraction was shown to reflect CSF, with pT181, pT217, and pT231 among the most prominent species identified in AD [
110]. A doubly-phosphorylated tryptic peptide (pT231 + pS235) specific of AD brain was identified in higher amounts than the monophosphorylated (pT231) counterpart [
110]. Using sarkosyl-insoluble samples, Wesseling et al. [
70] performed a quantitative and qualitative tau protein profiling (FLEXITau and Q-Exactive MS) of 29 AD patients and 28 matched control individuals. The study demonstrated that the abundance of insoluble tau is higher in AD than in healthy controls, with pathogenic tau aggregates predominately composed of the 0N, 1N, and 4R isoforms. The most relevant phosphosites specific to AD were pS199, pS202, and pT205 [
70]. The total number of phosphorylated residues identified by all methods in normal or AD/CBD human brain tissue stands at 56, representing well over half of all hydroxyl amino acids in tau [
70] (
Figure 1A).
Tau acetylation (identified on 21 lysines) directly contributes to the accumulation of phosphorylated tau, affecting tau turnover in CBD and AD [
70,
86,
111] (
Figure 1A). Acetylation at K281, K331, K343, and K353 of tau fibrils from CBD patients’ brains and at K298, K311, K331, K343, K353, and K369 from AD patients was described thanks to Orbitrap MS [
86]. Within the tau proline-rich domain, K163, K174, and/or, K180 have been reported as acetylation sites by immunoprecipitation and MALDI-TOF MS, with occupancy detectable in a normal brain and increasing with Braak stage in AD brain [
112].
Tau has the highest number of ubiquitination sites (17 sites) per any protein in AD, as identified by Orbitrap MS [
32,
70,
101]. Tau is almost exclusively ubiquitinated and acetylated in the tandem repeats R1-R4 and K369-E380 of sarkosyl-insoluble fractions from CBD and AD post-mortem tissue [
86]. Within these regions, ubiquitination can also occur at different sites of tau for AD (K254, K259, K267, K311, K317, K321) and CBD (K254, K343, K369, K375) [
86] (
Figure 1A). Recently, a structure-based model in which specific ubiquitination of tau influences the resulting filamentary structure was built by combining results from LC-MS analysis and cryo-electron microscopy observations using CBD patients’ brains [
86].
Tau methylation is a relatively recent discovery with several lysine residues being methylated in AD patient brains (4 sites: K67, K87, R406, K438) [
70,
113] and CBD (2 sites: K132, R349) [
86], as shown by Orbitrap MS (
Figure 1A). Interestingly, tau methylation appeared distributed among at least 11 sites, in the form of mono- and dimethyl lysine residues, primarily focused on the microtubule-binding repeat region, in four cognitively normal human brains using Orbitrap MS [
114].
High mannose-type sugar chains and truncated
N-glycans were found on tau in addition to a small amount of sialylated bi- and tri-antennary sugar chains. Truncated glycans were found richer in AD paired helical filaments (PHF)-tau than in AD cytosolic phosphorylated tau, which has been suspected of promoting the assembly and/or the stability of the pathological fibrils in AD [
87]. In addition, tau can be glycosylated by
O-GlcNAc (
Figure 1A), which is inversely proportional to the amount of phosphorylation [
115,
116] and responsible for slowing down neurodegeneration and preventing aggregation [
117]. Confirmation of these observations requires further investigation of the relative abundance of tau PTMs and isoforms in the brain, then in CSF, and ideally plasma, for biomarker discovery.
CSF. Quantitative high-resolution MS/MS strategies, including PRM, have given new insights into tau metabolism and truncation [
118,
119]. Specifically, these highlight differences in the relative abundance of PTMs between the brain and CSF tau of AD patients, notably, differences in truncations of the C-terminus [
119] and opposite trends in phosphorylation rate depending on the sites. A recent publication also identified site-specific phosphorylation changes in CSF along with the AD progression, particularly a reversal of pT181 at the onset of cognitive decline, which suggests sequestration in the brain of specific tau species [
88]. These recent observations raised the question of the relationship between CSF and brain tau. In the CSF, detection of soluble pT181, pT217, and truncated tau forms (e.g., Tau368) have been investigated for differential diagnosis and distinguish AD from other dementias using an Orbitrap MS [
24,
88]. The release of pT217 from the brain to CSF was linked to increased phosphorylation in AD. CSF pT217 was more accurate in detecting the presence of amyloid plaques (identified by PET) than other sites, such as pT181 [
24,
120] and pT205 [
88]. Remarkably, increased CSF levels of pT217 are closely related to amyloid plaques at the asymptomatic stage [
121]. Phosphorylation occupancy on pT217 is also lower intracellularly in the brain than extracellularly in CSF [
67]. Phosphorylation on pT205 and pS208 was detected in the CSF but not in the brain from healthy controls [
67]. Interestingly, the benefit of combining tau and α-synuclein for differentiation of DLB, AD, and controls was recently suggested [
122].
Blood/Serum. Brain and CSF proteins are transferred to the blood through the blood-brain barrier, arachnoid granulations, and the glymphatic system [
123]. Different studies have demonstrated that disease-associated protein alterations in the brain can be detected in the blood. A similar tau C-terminal truncation pattern in plasma compared to CSF was reported, with 15 tau peptides from residues 6–254 being detected, including 0N, 1N, 2N, and 3R-specific peptides using Orbitrap MS [
85,
118,
119,
124]. An inferred abundance of 0N/1N/2N peptides indicated similar contributions to previous reports in the brain and CSF (∼5/5/1) [
119]. pT217 and pT181 in plasma were highly specific for amyloid plaque pathology [
85]. However, there is strong evidence of a non-brain peripheral contribution of tau in plasma, with a different phosphorylation profile than CSF [
89]. Findings support blood phosphorylated tau isoforms (pT181, pT217, and pT231) as potentially helpful in detecting AD pathology, staging the disease, and diagnosis [
89].
3.2.2. α-Synuclein
α-synuclein is widely distributed in the CNS and is involved in the packaging, trafficking of vesicles, and regulation of synaptic plasticity [
125]. For reasons not fully comprehended, α-synuclein is prone to misfolding and forming fibrillar and aggregated forms within Lewy bodies and Lewy neurites-a typical pathological hallmark of PD and other synucleinopathies [
126]. The molecular factors triggering α-synuclein aggregation and Lewy bodies formation remain unknown. Changes in α-synuclein phosphorylation could represent a response to biochemical events associated with PD pathogenesis [
127].
Brain tissues. α-synuclein within Lewy bodies is phosphorylated at S87, S129, or Y125 as deduced from MS data in PD [
90,
128]. Phosphorylated α-synuclein at S129 accounts for more than 90% of α-synuclein found in Lewy bodies [
129]. In contrast, only 4% or less of total α-synuclein is phosphorylated at this residue in the normal brain [
91,
128]. This suggests that the accumulation of S129-phosphorylated α-synuclein is somehow related to the formation of Lewy bodies and dopaminergic neurodegeneration in PD [
90,
128]. Ubiquitination (K12, K21, and K23, in phosphorylated α-synuclein forms) is only detected in Lewy bodies [
91]. Together with truncation, these ubiquitinated and phosphorylated forms are present in the detergent-insoluble fraction of the familial PD brain (synuclein A53T mutation) [
91]. Several other α-synuclein proteoforms, including accumulation of truncated forms at C-terminal (Ac-α-syn
1–119) and N-terminal (α-syn
71–140, α-syn
68–140, α-syn
66–140, α-syn
65–140), mainly in the cingulate cortex, were described in PD [
130]. The levels of α-synuclein forms in Lewy body-enriched α-synuclein fraction (α-syn
1–6, α-syn
13–21, α-syn
35–43, α-syn
46–58, α-syn
61–80, and α-syn
81–96) were significantly increased in the PD cingulate region compared to controls [
92]. In addition, brain-derived α-synuclein is mainly N-terminally acetylated in Lewy body-enriched PD brain tissue fractions, as characterized by intact protein LC-Orbitrap MS [
92] (
Figure 1B). Although phosphorylation, acetylation, ubiquitination, and truncation may play an important role in α-synuclein biology, our understanding of the precise effects of these modifications in the biology and pathophysiology of neurodegenerative diseases is still partial. Of note, deposits of α-synuclein and S129-phosphorylated α-synuclein in the brain could be detected in people with non-neurodegenerative disorders [
22].
CSF/Blood/Serum. Monomeric, oligomeric, and post-translationally modified α-synuclein can be detected in body fluids such as CSF, plasma, and red blood cells [
131,
132,
133]. When analyzing α-synuclein as a biomarker, the high concentration in red blood cells should be considered and requires monitoring of blood contamination in CSF [
122]. Mass spectrometry using MRM mode showed CSF synuclein concentrations 550% superior to ELISA but similar levels in serum of subjects without neurodegenerative diseases [
133]. This difference could indicate the presence of different α-synuclein species in serum and CSF. CSF synucleins (alpha, beta, and gamma) were detected as being increased by MRM in AD and CJD, but no alteration was detected for synucleinopathies (PD, PD dementia (PDD), DLB) [
133]. Moreover, α-synuclein peptides α-syn
61–80 and α-syn
81–96 from the non-Abeta component (NAC) region have a 38 and 40% lower concentration in CSF than α-syn
24–32 from the N-terminal region [
133]. In 2020, the same group found an increased β-synuclein in patients with mild cognitive impairment and CJD, but not for PD [
134]. pS129 and ubiquitination (at multiple sites) have been detected in the CSF and plasma of PD, MSA, and DLB cases [
93]. In red blood cells, Pero-Gascon et al. [
135] detected N-acetylated α-syn as the main proteoform in healthy individuals and some PD patients (stage III and IV). Yang et al., 2017 found that α-syn
81–96, TVEGAGSIAAATGFVK, was highly correlated with disease severity and allowed the tracking of the PD progression [
94].
3.2.3. Amyloid Precursor Protein (APP) and Amyloid-Beta Peptides
The function of amyloid precursor protein (APP) in the body is unknown; however, evidence suggests its role in synaptic formation and function, neuronal outgrowth, protein trafficking, signal transduction, and intracellular calcium homeostasis. In pathological conditions, the proteolytic processing of APP by the β and γ secretase, which cleaves the protein at specific sites, generates Aβ peptide fragments with variable lengths of 37–43 amino acids. Among them, Aβ42 peptides are more prone to aggregation [
136]. In the brain, Aβ42
6–16 and Aβ42
17–28 (APP695 region 597–638) showed significant increases in asymptomatic AD and AD groups compared with control samples [
31]. The Aβ42/Aβ40 ratio in plasma was measured with LC-MS in AD and mild cognitive impairment patients to see its correlation to amyloid PET status and was shown to be a good predictor of Aβ PET positivity [
95]. Using combined immunoprecipitation and HR-MS techniques, it was shown that the relative levels of Aβ16 in AD compared to controls are increased in CSF [
96]. It was identified in AD 37 APP/Aβ glycopeptides with sialylated core-1 like
O-glycans attached to Thr(−39, −21, −20, and −13) in a series of APP/AβX–15 glycopeptides, where X was −63, −57, −52, and −45, concerning Asp1 of the Aβ sequence [
97]. An increase in Y10
O-glycosylated Aβ peptides linked to (Neu5Ac)
1–2Hex(Neu5Ac)HexNAc-O-structures was observed in CSF in six AD patients compared to seven non-AD patients [
97]. Short Aβ isoforms (N-terminal −3, 1, 4, 5; C-terminal 15, 16, 17, 18, 19, 20) were detected with
O-glycosylation at Y10 of Aβ [
97,
137] (
Figure 1C). Despite this, further investigations are warranted to address the significance of glycosylation in APP proteolysis and its consequences on amyloid deposition.